
# Small Variations
# Calling
The SEQ platform has standard analysis versions pre-setup for every kit defined in the system. Calling variants are performed differently in different analysis versions.
# Analysis versions for Tumor/Normal Matched Samples
| Name | Explanation | Alignment/ variant calling | BAM processing | Primer Trimming | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|
| Sentieon BWA-TNhaplotyper2-PCR Dedup- somatic | Optimized for Exome Samples. | Sentieon BWA / TNhaplotyper2 | PCR Dedup | N/A | hg38,hg19 | Illumina, MGI |
| Sentieon BWA-TNScope-PCR Dedup- somatic | Optimized for capture-based somatic kits. | Sentieon BWA / TNScope | PCR Dedup | N/A | hg38,hg19 | Illumina, MGI |
# Analysis versions for Tumor Only Samples
| Name | Explanation | Alignment/ variant calling | BAM processing | Primer Trimming | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|
| BWA-Freebayes-PCR Dedup-Indel Realignment- somatic | Optimized for capture-based somatic kits. | BWA / Freebayes | PCR Dedup + Indel Realignment | N/A | hg38,hg19 | Illumina, MGI |
| BWA-Freebayes-BamKeser-Indel Realignment-Long Indel Finder- somatic | Optimized for amplicon-based somatic kits. Performs an additional step for long indel alterations. | BWA / Freebayes | Indel Realignment | BamKeser | hg38,hg19 | Illumina, MGI |
| BWA-Freebayes-BamKeser-Indel Realignment- somatic | Optimized for amplicon based somatic kits. | BWA / Freebayes | Indel Realignment | BamKeser | hg38,hg19 | Illumina, MGI |
**BamKeser is our in-house designed and precisely working primer trimming tool.
After variant calling, the Genomize SEQ platform processes the resulting VCF file to form a Genomize standard VCF file, which can be downloaded through the platform. The Genomize standard VCF line will have gstd=1 in the info field. Standardization of the VCF file includes the following important steps:
- Minimal variant representation: Some callers produce redundant bases at the left-hand or right-hand side of either alternative or reference allele. This redundancy has to be removed to obtain the correct annotation of variants in the subsequent steps.
- Multi nucleotide polymorphism decomposition: MNPs are decomposed into all possible smaller versions of that variation. This is critical not to miss the annotation of a possible variant in the sample.
# Annotation
Genomize standard VCFs are annotated by using the VEP (opens new window) tool from NCBI, which is a trusted and constantly developed open-source annotation tool. Computational prediction algorithms are provided by VEP includes sift, polyphen, mutationtaster, revel, metalR, and DANN.
In addition, the variants are annotated with the dbSNP database and the ClinVar database.
# Extended Annotation
Genomize offers the "Extended Annotation Service" to all SEQ Platform users as a part of its basic services. With the "Extended Annotation Service", our users are able to see the variants in the non-reference Ensembl and RefSeq transcripts in the variant list in case these variants increase the pathogenicity. With this functionality, it is now even less likely to "miss" a variant using the SEQ Platform.
The variants listed thanks to the "Extended Annotation Service" are marked with the  icon in the variant list.
icon in the variant list.
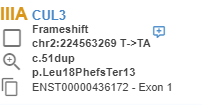
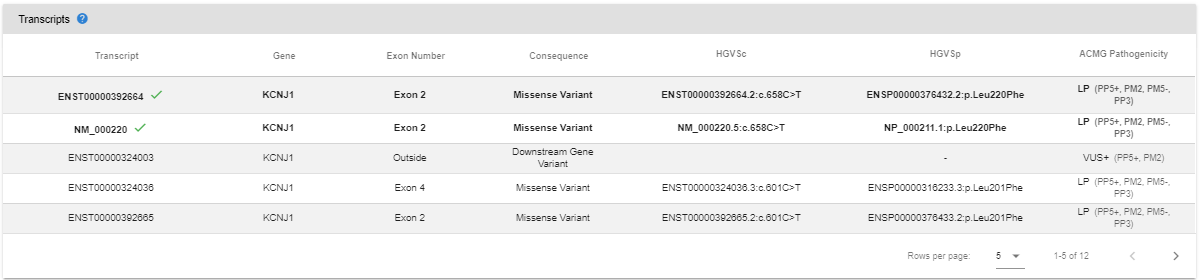
You can also see the ACMG pathogenicity predictions for all known transcripts on the variant page.
# Visualization
# Small Variants Tab
The information related to a variant can be visualized in the "Variants" tab.
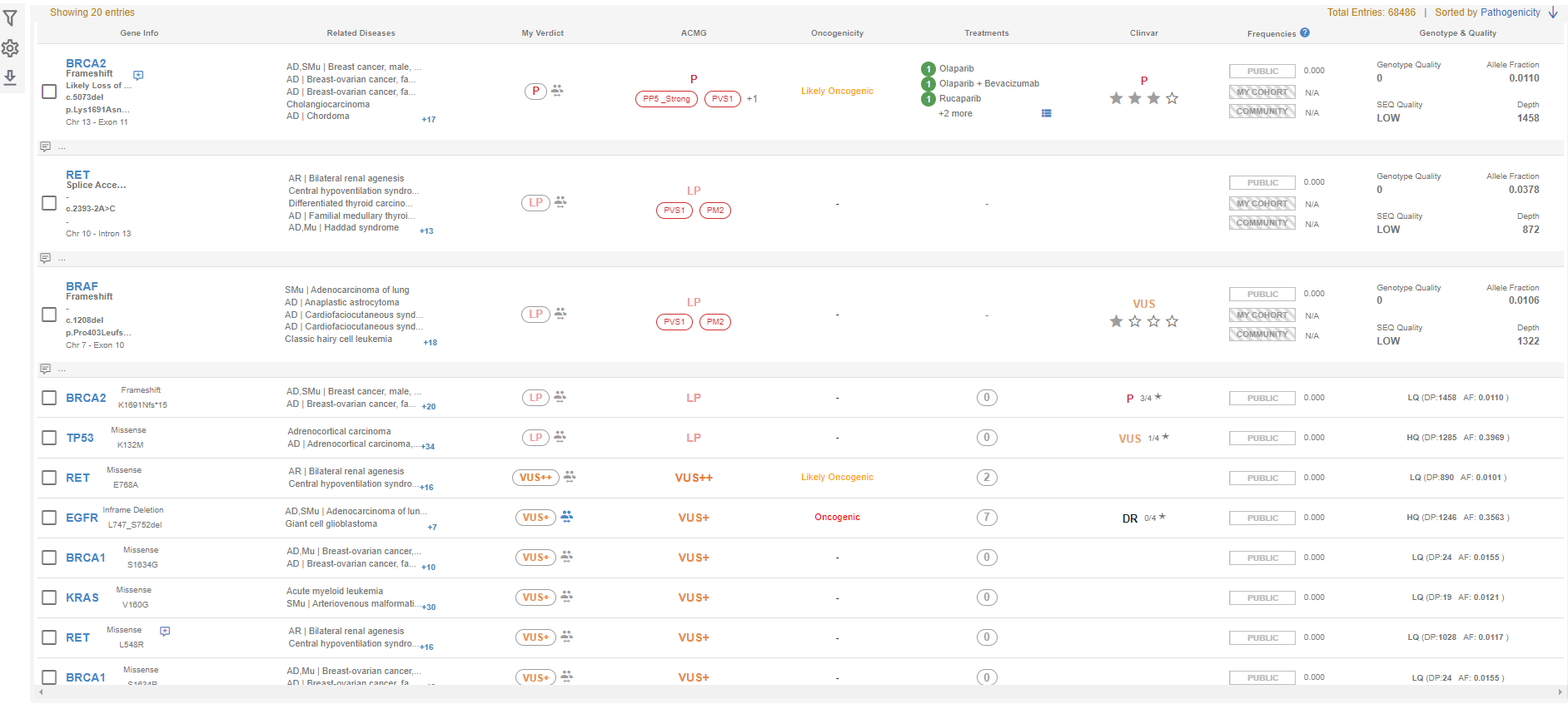
Here, you will see the information required for variant evaluation in various columns as well as more detailed information about the gene and the variant in the summary area at the bottom of the page.
- Gene Info: This column will list the gene name, the effect of the variant on the protein, the change in the transcript and the protein, as well as the chromosome of the gene and the affected exon.
- Related Diseases: The name and MOI of the disease(s) associated with the gene are listed here. If there are more than 5 entries, you will see a "+x" icon at the bottom right to give you the total number of listed diseases. You can mouse over the list to see the complete list.
- My Verdict: The SEQ Platform allows you to override the ACMG prediction. You can change your own ACMG classification by clicking on the "ACMG classification" in this column. If you change the classification, this change will be saved in your center's database, and you and your colleagues will see your choice in your future samples. You will also see a "people" icon on the right of this field. If any other center in the SEQ community makes a change in the ACMG classification of this variant, you will be able to see a concise list of the number of other centers that have changed the pathogenicity of this variant, and to which classification.
- ACMG: Here, you will see Genomize's ACMG classification for the variant as well as the assigned evidence codes.
- Oncogenicity: You will see the oncogenicity status of the variant in this column.
- Treatments: You will see the available treatments (if any) in this column. You can mouse over the treatment option to see the detailed scientific information as well as literature references.
- ClinVar: This column shows the ClinVar entries and the star status of the variant.
- Frequencies: The frequency of the variant in "public" databases (i.e. GnomAD, ESP6500, UK10K, etc.), in your patient cohort, and the SEQ Community cohort, respectively. You will also see a "diverged arrow" icon if there is an equivalent variant in the hg19 genome version. When you mouse over this icon, you will see the equivalent variant's frequency information as well.
- Genotype and Quality: Here, you will see the visual representation of the variant's genotype as well as various quality information. The compound heterozygous variants are represented with a "shadowed" chromosome image. Clicking on the chromosome image will list the compound heterozygous variants in the sample.
- Genotype Quality: The quality value assigned by the variant caller used in your analysis.
- SEQ Quality: If the number of reads of the variant is between the primary and secondary coverage thresholds (5 and 50 by default, respectively), the variant will be classified as LOW quality. If it is higher than the secondary coverage threshold, it will be classified as HIGH quality. The threshold values can be changed during the sample upload or through the "Site settings".
- Allele Fraction: The ratio of the reads containing the variant to all reads covering the variant's position.
- Depth: The total number of reads covering the variant's position.
# Information Field
When you click on a variant card, the information field will pop up at the bottom of the page. In this field, you will see several tabs:
- Summary: This tab contains summary gene & variant information, a radar plot visualizing the pathogenicity and the relevance of the variant according to 5 categories, detailed information about the variant's classification in Genomize's AI-assisted variant prioritization feature, and external links for the variant and/or the gene.
- Frequencies: Here, you will see the frequency tables for the variant within your cohort, the SEQ community cohort, and the public databases:
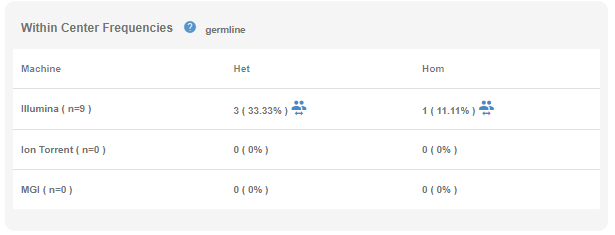
- Within Center Frequencies: In this table, you will see the frequency values as well as the incidences of the variant among your own sample cohort stratified by the sequencing platform and genotype. If the variant is encountered in other samples, you will see a "people" icon next to the frequency information. By clicking this icon, you can view the list of samples that carry this variant, and click on the sample name to open the sample in a new tab.
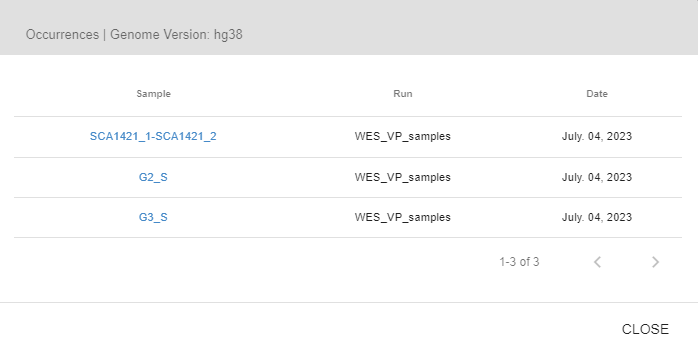
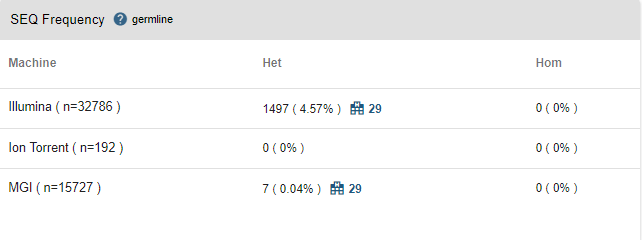
- SEQ Frequency: In this table, you will see the same information but for the whole SEQ community. Joining the SEQ community is optional and you can opt out anytime you like. If the variant has been encountered by any other center in the SEQ community, you will see a "hospital" icon next to the frequency information. You can see the number of other centers, and by clicking on this icon, you can get in contact with the other centers. You will not be able to obtain any information about the name of the center and the recipient unless the receiving party accepts your request.
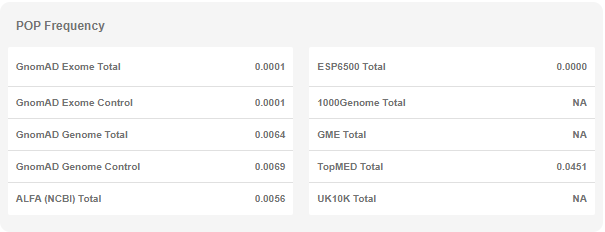
- Population Frequencies: This table shows the summary frequency values in various public databases. You can see the stratified values for each database in the "Variant Page" (see below).
- Similar Cases: In this tab, you will see the phenotypes observed in samples that carry this variant. The phenotype does not need to be observed in the sample you are currently analyzing. You can also see the number of samples with the specified phenotype as well as the names and links to those samples.
- ACMG: Here, you can obtain more information about the ACMG classification of the variant. You can also use this tab as an ACMG calculator by adding, removing, or modifying the evidence codes.
- ClinVar: In this tab, you will see the ClinVar entries for the variant and relevant links.
- Predictions: Here you will see prediction scores from various in-silico tools for pathogenicity, evolutionary conservation, and splicing.
- Validation: This form can be used to keep track of verifications (ex: Sanger or qPCR) for the variant.
- Literature: You will see the list of publications for the variant and the gene listed separately. You can toggle the "disease-related only" switch to list all publications or only disease-related publications. This literature search is performed at the time of sample upload.
- Isoforms: All transcript annotations for the variant are listed in this table. The reference transcripts will appear on top with the default sorting. Please see the "Extended Annotation" section for more details.
# Filtering
The SEQ Platform offers various filtering options. You can open the filter options by clicking the "funnel" icon located at the top-left of the page.
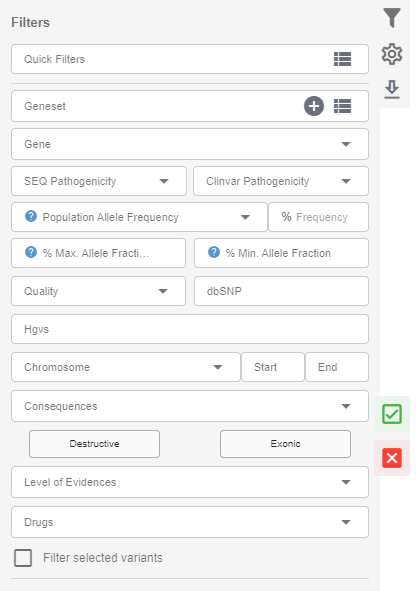
In the filters field, you will see the following:
- Quick filters: You can select a filterset you previously created for a quick application. You can create a new filterset by clicking the icon on the right of the "Quick Filters" section.
- Geneset: You can create your own geneset using the plus icon in this field and use it in your filters. You can create as many genesets as you need. You can also use the system-defined genesets, which are updated weekly, or as the new version is released in the case of ACMG's list of recommended genes for incidental findings.
- Gene: You can search for variants in the gene(s) you want in using this option. You can search for multiple genes.
- SEQ pathogenicity: You can select the ACMG classification by the SEQ Plaform to filter for variants in this section.
- ClinVar pathogenicity: You can use ClinVar's variant classification categories for your filtering in this section.
- Population allele frequency: You can search for variants with higher/lower fractions in the public databases than the value you provide here. The population frequency filter will consider the GnomAD exome frequencies if the variant is covered in the GnomAD. The maximum allele frequency from the Exac, Esp6500, and 1000genome total frequencies will be used only if the variant is not covered in GnomAD. The reason for this order is due to the presence of possible ethnicity biases in the Esp6500, Exac, and 1000genome which are less likely to be present in the GnomAD since GnomAD contains many more samples from an evenly distributed sample set in terms of ethnicity. For instance, some well-established cystic fibrosis variants are present in more than 5% of the ESP6500 EA population.
- Quality: You can filter for high- or low-quality variants. Please see above for the "SEQ Quality".
- % Min. allele fraction & % max. allele fraction: You can provide % values in these fields to filter for variants within the range of these values.
- Chromosome, start and end: You can search for variants within the genomic coordinates you provide in this section.
- dbSNP: You can search for a specific variant using the rdID here.
- HGVS: You can search for a specific variant using the HGVS identifier here.
- Consequences: You can select the effect of the variant on the protein/transcript here. You will see two shortcut buttons for "Destructive" and "Exonic" effects.
- Level of Evidences: You can filter for the variants with treatment options using various categories in this field.
- Drugs: You can filter the variants using the drug information.
- Filter selected variants: If an option is selected, only the variants that have the "report checkbox" ticked will be used for filtering.
Please note that the filters work with the "AND" operator.