# Data Upload
With the SEQ Platform, you can directly upload your files or use the cloud browser to browse and select the files pre-uploaded to your account.
# Direct Data Upload
Click on the "Upload" button at your homepage and select "Germline" option. Here, you can select FASTQ, gVCF or VCF options.
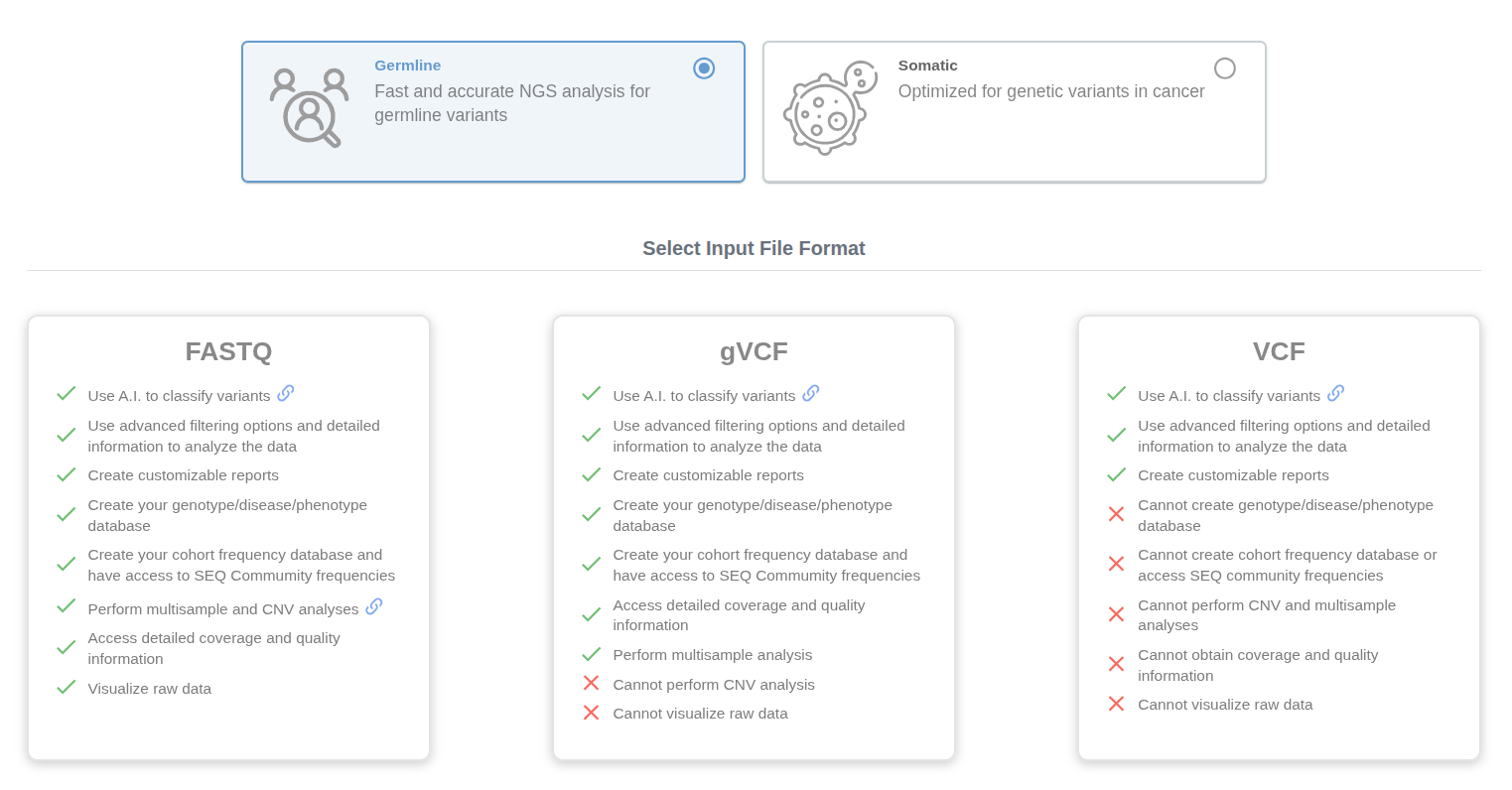
# FASTQ Upload
# Run Selection
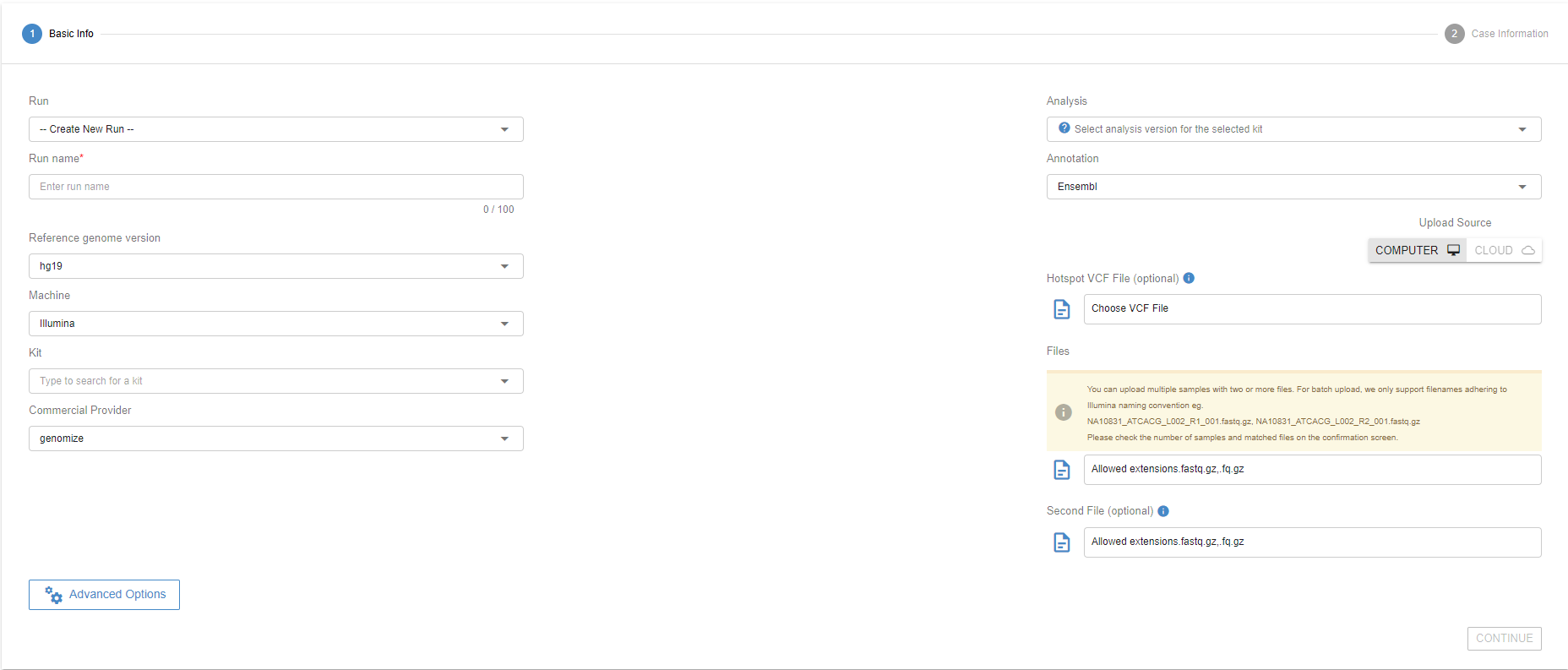
You can upload your samples to a new run by selecting “Create New Run” under “Run Name” and giving it a name on the “Name For New Run” field. You can also upload your samples to an existing run by selecting the previous run from the dropdown menu.
Run information is very critical for Copy Number Variation analyses. Therefore, please make sure you organize the samples under the runs in the same way as you process your sample materials. Ideally, a run is a set of samples coming from the same wet-lab and run process (flow cell, etc.).
# Choose the technology type
Choose the next-generation sequencing machine associated with the samples. If you do not know which sequencing platform is used, you can select the "Unknown" option. Mixing different technologies in one run is not permitted.
# Choose the kit type
The SEQ platform has hundreds of different kits predefined in the system. A new kit can be defined with a set of target coordinates and a list of targeted genes. The Addition of a new kit typically takes one business day. For kit requests, please contact us through [email protected].
Every kit is associated with a standardized analysis version in SEQ. Probe-based kits, primer-based kits, Illumina & MGI technology, ION torrent technology, germline analysis, or somatic analyses all have a preset analysis version.
# Select the files to upload
Click the "Browse" button under the “File” to upload all the files you want to analyze. Make sure that you upload both read files for paired-end reads. See the table below for the supported input file types:
| File Types | Batch Sample Upload | |
|---|---|---|
| Illumina | .fastq.gz, .fq.gz | Supported |
| Ion Torrent | .bam | Supported |
| MGI | .fastq.gz, .fq.gz | Supported |
| Pacific Biosciences | .bam | Supported |
| Oxford Nanopore Technologies | .fastq.gz, .fq.gz | Supported |
| Element Biosciences | .fastq.gz, .fq.gz | Supported |
| GeneMind | .fastq.gz, .fq.gz | Supported |
| Onso (PacBio) | .fastq.gz, .fq.gz | Supported |
| Salus | .fastq.gz, .fq.gz | Supported |
# Filename formatting for batch upload
# Illumina
You can upload multiple samples with two or more files. For batch uploads, we only support filenames following the “Illumina naming convention” e.g.
NA10831_ATCACG_L002_R1_001.fastq.gz
NA10831_ATCACG_L002_R2_001.fastq.gz
The filenames from Illumina platform are handled as below:
<name_field1>_<name_field2>_<lane_#>_<read_#>_<always_001>.fastq.gz
Name field 1, name field 2, lane #, and read # are used to match the corresponding files correctly.
You can alter the name fields 1 and 2 without using space, punctuation, or underscore (_) characters.
Following this naming convention, you can upload multiple samples (each with multiple fastq.gz files).
The --no-lane-splitting parameter must not be used during file conversion with bcl2fastq, DRAGEN BCL Convert, or similar tools.
Lane identifiers are required to remain present in FASTQ file names for correct file matching. Please refer to the tools’ documentations for details.
Please check the number of samples and matched files on the confirmation screen.
# MGI
You can upload multiple samples with two or more files. For batch uploads, we only support filenames following the “MGI naming convention” e.g.
V12345678_L01_16_1.fastq.gz
V12345678_L01_16_2.fastq.gz
The filenames from MGI platform are handled as below:
<flowcell_id>_<lane_#>_<barcode>_<read_#>.fq.gz
Flowcell ID, lane #, barcode, and read # are used to match the corresponding files correctly.
You can alter the flowcell ID field without using space, punctuation or underscore (_) characters.
If you have used more than one barcode for the same sample, you need to rename the file as follows:
Original file names:
sample1234_L01_16_1.fq.gz
sample1234_L01_16_2.fq.gz
sample1234_L01_17_1.fq.gz
sample1234_L01_17_2.fq.gz
Altered file names:
sample1234_L01_16_1.fq.gz
sample1234_L01_16_2.fq.gz
sample1234_L02_16_1.fq.gz
sample1234_L02_16_2.fq.gz
In this example, barcode numbers of the last two files are changed to 16, and their lane numbers are increased by 1.
Please check the number of samples and matched files on the confirmation screen.
# IonTorrent
IonTorrent files need to be unaligned BAM files with .bam extension.
# Pacific Biosciences
Pacific Biosciences files need to be BAM files with .bam extension. You can upload multiple samples in a single batch. One file for each sample is expected.
# Oxford Nanopore Technologies
Oxford Nanopore Technologies files need to be in fastq.gz or fq.gz format. You can upload multiple samples in a single batch. One file for each sample is expected.
# Element Biosciences
You can upload multiple samples with two or more files. For batch uploads, we only support filenames following the “Element Biosciences naming convention” e.g.
SampleName_S1_L001_R1_001.fastq.gz
SampleName_S1_L001_R1_001.fastq.gz
The filenames from Element platforms are handled as below:
<name_field1>_<name_field2>_<lane_#>_<read_#>_<always_001>.fastq.gz
Name field 1, name field 2, lane #, and read # are used to match the corresponding files correctly.
You can alter the name fields 1 and 2 without using space, punctuation, or underscore (_) characters.
Following this naming convention, you can upload multiple samples (each with multiple fastq.gz files).
Please make sure the Lane identifiers are present in the file names as they are required for correct file matching.
You can use --legacy-fastq parameter in Bases2Fastq to produce files with names adhering to the above format.
Please refer to the Bases2Fastq documentation for details.
Please check the number of samples and matched files on the confirmation screen.
# GeneMind
You can upload multiple samples with two or more files. For batch uploads, we only support filenames following the “GeneMind naming convention” e.g.
SampleName_R1.fq.gz
SampleName_R2.fq.gz
The filenames from GeneMind platform are handled as below:
<name_field>_<read_#>.fq.gz
Name field and read # are used to match the files correctly.
You can alter the name field without using space, punctuation and underscore (_) characters.
Following this naming convention, you can upload multiple samples (each with multiple fq.gz files).
Please check the number of samples and matched files on the confirmation screen.
# Salus
You can upload multiple samples with two or more files. For batch uploads, we only support filenames following the “Salus naming convention” e.g.
SampleID_R1.fastq.gz SampleID_R2.fastq.gz
The filenames from Salus platform are handled as below:
<sampleID>_<read_#>.fastq.gz
SampleID and read # are used to match the files correctly.
You can alter the sampleID field without using space, punctuation and underscore (_) characters.
Following this naming convention, you can upload multiple samples (each with multiple fastq.gz files).
Please check the number of samples and matched files on the confirmation screen.
# Onso (Pacific Biosciences)
You can upload multiple samples with two or more files. For batch uploads, we only support filenames following the “Obc2fastq v6.0 naming convention” e.g.
ABCD123_L01_R1_Sample_Information.fastq.gz
ABCD123_L01_R2_Sample_Information.fastq.gz
<Flowcell_id>_<LaneSpec>_<ReadSpec|IndexSpec>_<Sample_id>.fastq.gz
FlowcellID, SampleID and ReadSpec|IndexSpec are used to match the files correctly. You can alter the FlowcellID and sampleID fields without using space or punctuation. Following this naming convention, you can upload multiple samples (each with multiple fastq.gz files).
Please check the number of samples and matched files on the confirmation screen.
# Hotspot VCF file upload
A predefined VCF file can be uploaded to SEQ. The VCF4.2 standard is supported. If the user does not upload a VCF as a hotspot, SEQ automatically subsets Pathogenic or Likely Pathogenic variants from ClinVar as the default hotspot for panels. For Whole Exome Sequencing, the default hotspot assignment is currently not supported.
# Advanced options
# Variant Calling parameters
A set of parameters is used to assess the quality of every variant called in a sample. Two parameters, the primary coverage threshold and the minimum alternative fraction threshold, can cause the classification of the variant as “FAILED”. The “FAILED” variant calls will not be displayed.
The variant calls with an alternative allele count less than the primary coverage threshold will be classified as “FAILED” and not be displayed.
The variant calls with alternative allele frequency less than the allele fraction threshold will be classified as “FAILED” and not be displayed.
# Other parameters
When calculating coverage metrics for the gene coverage and the kit’s on-target coverage percentages, SEQ uses four different thresholds. 1X and 5X are the preset values. The other two values may be customized by the user per upload.
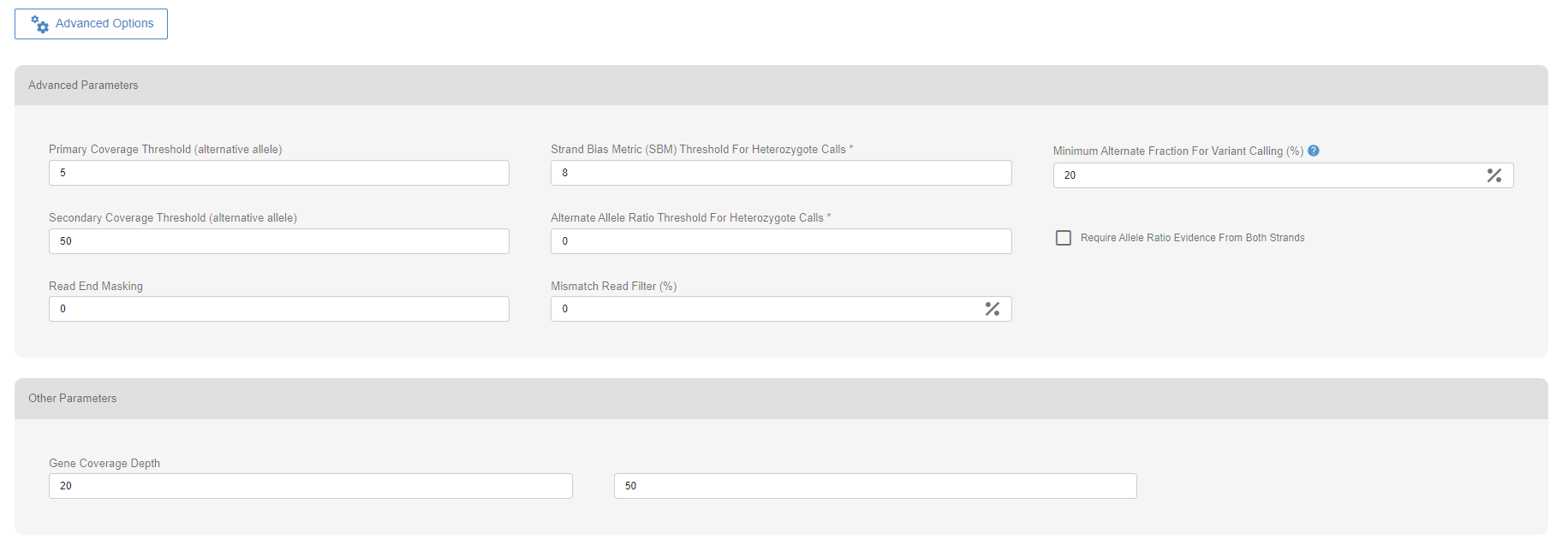
The default values of the advanced options are set under “Site Settings” in the Settings menu.
# Choose Analysis Version
SEQ has standard analysis versions pre-setup for every kit defined in the system. Data processing and variant calling are handled differently based on the sample type, sequencing platform, and selected analysis pipeline.
# Analysis versions for long-read WGS
| Name | Explanation | Alignment/ variant calling | CMRG Support* | SV Calling** | Mito Calling | Phasing | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|---|---|
| Sentieon minimap2-DNAScope-SV Calling-Phasing-germline | Optimized for ONT long-read WGS samples | Sentieon minimap2 / DNAScope | No | Sentieon LongreadSV | — | Variant Phaser DNAscopeHP | hg38 | Oxford Nanopore |
| PacBio pbmm2-DeepVariant-SV calling-Phasing-germline | Optimized for PacBio long-read WGS samples | PacBio pbmm2 / DeepVariant | ***Paraphase | Sawfish, TRGT | Mitorsaw | HiPhase | hg38 | PacBio |
* CMRG: Challenging Medically Relevant Genes (Wagner et al., 2022 (opens new window))
** Structural Variant Calling: See Structural Variant Detection for more details.
*** Paraphase: PacBio's recommended tool for detecting segmental duplication regions and medically relevant genes, including SMN1/SMN2, HBA1/HBA2, GBA, CYP21A2, CYP2D6, CYP11B1, OPN1LW, PMS2, STRC, NEB, IKBKG, and NCF1 (Chen et al., 2025 (opens new window)). See Targeted Variant Calling for more details.
# Analysis versions for short-read WGS
| Name | Explanation | Alignment/ variant calling | BAM processing | SV Calling | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|
| Sentieon BWA-DNAScope-SV Calling-germline | Optimized for WGS samples prepared with a PCR enrichment step | Sentieon BWA / DNAScope | MarkDuplicate | Delly, Manta, Tiddit, ExpansionHunter | hg19, hg38 | Illumina MGI Element Salus |
| Sentieon BWA (PCRfree)-DNAScope-SV Calling-germline | Optimized for WGS samples prepared without a PCR enrichment step | Sentieon BWA / DNAScope | MarkDuplicate | Delly, Manta, Tiddit, ExpansionHunter | hg19, hg38 | Illumina MGI Element Salus |
| BWA-Freebayes-SV Calling-germline | Optimized for WGS samples | BWA / Freebayes | PCR Dedup + Indel Realignment | Delly, Manta, Tiddit, ExpansionHunter | hg19, hg38 | Illumina MGI Element GeneMind Onso Salus |
| BWA-GATK-SV Calling-germline | Optimized for WGS samples | BWA / GATK | PCR Dedup + Indel Realignment | Delly, Manta, Tiddit, ExpansionHunter | hg19, hg38 | Illumina MGI Element GeneMind Onso Salus |
# Analysis versions for capture based targeted panels, including WES
| Name | Explanation | Alignment/ variant calling** | BAM processing | CNV Calling (Cohort Mode) | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|
| Sentieon BWA-DNAScope- germline | Optimized for capture-based germline kits. | Sentieon BWA / DNAScope | MarkDuplicate | GATK-CNV + delly* | hg19, hg38 | Illumina MGI Element GeneMind Onso Salus |
| BWA-Freebayes-PCR dedup - germline | Optimized for capture-based germline kits. | BWA / Freebayes | PCR Dedup | GATK-CNV + delly* | hg19, hg38 | Illumina MGI Element GeneMind Onso Salus |
| BWA-Freebayes-PCR dedup-Indel Realignment - germline | Optimized for capture-based germline kits. Default analysis for most kits. | BWA / Freebayes | PCR Dedup + Indel Realignment | GATK-CNV + delly* | hg19, hg38 | Illumina MGI Element GeneMind Onso Salus |
| BWA-GATK-PCR dedup-Indel Realignment - germline | Optimized for capture-based germline kits. Uses a GATK variant caller. | BWA / GATK | PCR Dedup + Indel Realignment | GATK-CNV + delly* | hg19, hg38 | Illumina MGI Element GeneMind Onso Salus |
| BWA-Freebayes High Sensitivity-PCR dedup-Indel Realignment - germline | Optimized for capture-based germline kits to call variants with a low fraction (<20%). | BWA / Freebayes | PCR Dedup + Indel Realignment | GATK-CNV + delly* | hg19, hg38 | Illumina MGI Element GeneMind Onso Salus |
* Delly is utilized only for panels with more than 100 genes.
** Mitochondrial analysis is performed following GATK best practices, and gnomAD filters are applied (for details, see Mitochondrial Variant Detection).
# Analysis versions for amplicon based panels
| Name | Explanation | Alignment/ variant calling | BAM processing | Primer Trimming | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|
| BWA-Freebayes-BamKeser-Indel Realignment - germline | Optimized for amplicon-based germline kits. | BWA / Freebayes | Indel Realignment | BamKeser | hg19, hg38 | Illumina MGI Element GeneMind Onso Salus |
| BWA-Freebayes-BamKeser - germline | Optimized for amplicon-based germline kits. Does not perform indel realignment step. | BWA / Freebayes | NA | BamKeser | hg19, hg38 | Illumina MGI Element GeneMind Onso Salus |
**BamKeser is our in-house designed and precisely working primer trimming tool.
# Analyis versions for IonTorrent uBAM samples
| Name | Explanation | Alignment/ variant calling | BAM processing | Primer Trimming | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|
| Torrent Suite 5.8 - No Trimming - Default Parameters v2 - germline | Optimized for IonTorrent samples. Does not perform primer trimming. | Torrent Suite 5.8 | NA | NA | hg19 | IonTorrent |
| Torrent Suite 5.8 - No Trimming - BRCA specific - germline | Optimized for IonTorrent BRCA samples. | Torrent Suite 5.8 | NA | NA | hg19 | IonTorrent |
| Torrent Suite 5.8 - No Trimming - CFTR specific - germline | Optimized for IonTorrent CFTR samples. | Torrent Suite 5.8 | NA | NA | hg19 | IonTorrent |
**BamKeser is our in-house designed and precisely working primer trimming tool.
# Submit your data
As the last step, you can upload your data by clicking the “Continue” button to start the upload process. After clicking "Continue", you will see the "Case Information" screen. Please refer to the "Genomize's Variant Prioritization" section for more information on entering the case information. You can then click the "Upload" button to see the number of analyses and the list of files matched for each analysis. Please be sure that both of these pieces of information are correct and hit "Approve" to start the upload process or "Cancel" to make changes.
When you start the upload, you will see the progress for each file. Transferred samples will immediately begin processing without waiting for the entire batch to finish uploading.
SEQ Platform's upload process is secure and performs a checksum to ensure the files are transferred correctly. Please do not close the browser tab or shut down your computer. Also, please ensure that your computer will not go into sleep/hibernation mode during the upload. Otherwise, the upload process will be aborted. Our upload process is resistant to intermittent loss of internet connection.
When the upload process is completed, you will be redirected to the corresponding Run's page, and your samples will be queued for analysis. Refresh the corresponding Run page to see the last status of the analysis.
# Small Variant Detection
The variant caller used depends on the analysis version selected during upload — see Choose Analysis Version for the full list of available pipelines per platform and kit type.
| Sample Type | Variant Caller |
|---|---|
| Short-read WGS | DNAScope, Freebayes, GATK |
| ONT Long-read WGS | DNAScope |
| PacBio Long-read WGS | DeepVariant |
| Capture-based panels (incl. WES) | DNAScope, Freebayes, GATK |
| Amplicon-based panels | Freebayes |
| IonTorrent | Torrent Suite |
For GATK and Sentieon (DNAScope) pipelines, a secondary IGV BAM track containing assembled haplotype data is also produced — see IGV BAM for details.
# IMPORTANT NOTE: POST-PROCESSING
After variant calling, the Genomize SEQ platform processes the resulting VCF file to form a Genomize standard VCF file, which can be downloaded through the platform. The Genomize standard VCF line will have GSTD=1 in the info field. Standardization of the VCF file includes the following important steps:
- Minimal variant representation: Some callers produce redundant bases at the left-hand or right-hand side of either alternative or reference allele. This redundancy has to be removed to obtain the correct annotation of variants in the subsequent steps.
# Mitochondrial Variant Detection
The revised Cambridge Reference Sequence (rCRS, NC_012920.1) is used as the reference for the mitochondrial genome across all platforms.
# Short-Read and Panel Samples
The mitochondrial variant calling pipeline follows GATK best practices (opens new window), using Mutect2 as the variant caller.
Filtering and genotype assignment for mitochondrial variants are performed in line with the recommendations from gnomAD (opens new window) . Briefly, SNVs with Variant Allele Frequency (VAF) below 0.01 were removed. Variants with VAF equal to or higher than 0.95 are classified as homoplasmic. Variants with VAF lower than 0.95 are classified as heteroplasmic. Any variants in previously reported artifact-prone sites (positions 301, 302, 310, 316, 3107, and 16182) are ignored.
In targeted panel analyses (including WES), mitochondrial variant detection is only available for panels with distinct chrM targets in their bed files.
In WGS analyses, mitochondrial variant detection uses the same variant callers and filtering steps as those applied to other chromosomal regions. If your WGS sample preparation is optimized for mitochondrial DNA isolation and enrichment, please contact support for further assistance.
# PacBio Long-Read WGS Samples
For PacBio HiFi long-read WGS samples, mitochondrial variant calling is performed by Mitorsaw (opens new window), a dedicated mitochondrial variant caller for HiFi data. Mitorsaw provides heteroplasmy detection with variant allele frequency (VAF) and heteroplasmy bounds (HPBOUND) fields.
# Structural Variant Detection
SEQ Platform can detect and report various structural variants listed below. For single samples, maximum SV size is limited to 100,000 bps****.
For cohort CNV analysis in WGS samples, please refer to Copy Number Variations section
# Supported Structural Variants
| SV-Group | Abbreviation | Supporting callers |
|---|---|---|
| Deletion | DEL | Manta, Tiddit, Delly, HiFiCNV, Paraphase, Sawfish |
| Duplication | DUP | Manta, Tiddit, Delly, HiFiCNV, Paraphase, Sawfish |
| Insertion | INS | Manta, Delly, Sawfish |
| Inversion | INV | Tiddit, Delly, Sawfish |
| Breakend (Unresolved)* | BND | Manta, Tiddit, Delly, Sawfish |
| Short Tandem Repeat** | STR | ExpansionHunter, TRGT |
| Complex*** | CPX | Manta, Tiddit, Delly, Sawfish |
* Structural variants that cannot be classified into any other type are listed as BND
** Only disease-associated STR variants are supported. For PacBio HiFi samples, the STRchive catalog (opens new window) is used; for short-read and ONT samples, the gnomAD STR catalog (opens new window) is used. See Special Note on Repeat Finding for details.
*** If more than one type of SV is detected in combination, they are classified as CPX variant. ex: DEL:INS, DUP:INV, etc. Currently only DEL:INS variants are supported.
# Variant callers used for SV detection
| Tool | Algorithm | Supported SV-types |
|---|---|---|
| Manta1 (opens new window) | Manta (opens new window) divides the SV and indel discovery process into two primary steps: 1. Scanning the genome to find SV associated regions. 2. Analysis, scoring and output of SVs found in these regions. | - Deletions - Duplications - Deletion-Insertions - Insertions - Breakends |
| Delly2 (opens new window) | DELLY (opens new window), short-range and long-range paired-end libraries are analyzed for discordantly mapped read pairs. Paired-end predicted structural variants are then refined using split-reads and reported at single-nucleotide breakpoint resolution. In addition to general parameters applied to SVs, insert size cutoff for split reads ≥ 15 bps, minimum paired-end MAPQ ≥ 20 filters are used for DELLY. | - Deletions - Duplications - Deletion-Insertions - Insertions - Inversions - Breakends |
| Tiddit3 (opens new window) | TIDDIT (opens new window), detects structural variants by examining sequences for discordant pairs, split reads, and supplementary alignments, which must exceed a specified quality threshold. It uses a clustering method similar to DBSCAN, where a cluster forms if sufficient signals are within a designated distance. Clusters lacking enough signals are discarded; otherwise, they are included in the output regardless of other quality filters. | - Deletions - Duplications - Inversions - Breakends |
| ExpansionHunter4 (opens new window) | ExpansionHunter (opens new window) is a tool designed for targeted genotyping of short tandem repeats (STRs) and flanking variants. It operates by analyzing BAM files to find reads that either span, flank, or are fully contained within each targeted repeat. This precise approach allows for effective characterization of these genomic elements, tailored specifically to identify and quantify repeat variations. | Short Tandem Repeats |
| HiFiCNV5 (opens new window) | HiFiCNV (opens new window) is a cutting-edge tool specifically designed for calling copy number variants (CNVs) using high-fidelity (HiFi) sequencing reads. It offers optimized segmentation and calling for germline whole genome sequencing (WGS) using HiFi reads, ensuring accurate results. The tool automatically estimates and corrects GC-bias, which enhances the reliability of the data. | - Deletions - Duplications |
| TRGT6 (opens new window) | TRGT (opens new window) is a tool for targeted genotyping of tandem repeats from PacBio HiFi data. In addition to the basic size genotyping, TRGT profiles sequence composition, mosaicism, and CpG methylation of each analyzed repeat and visualization of reads overlapping the repeats. For details on the repeat catalog, see Special Note on Repeat Finding. | Short Tandem Repeats |
| Paraphase7 (opens new window) | Paraphase (opens new window) is a Python tool that takes raw HiFi aligned BAMs as input (whole-genome or enrichment) before phasing, phases haplotypes for genes of the same family, determines copy numbers and makes phased variant calls with full haplotype resolution. Paraphase supports 160 segmental duplication regions (opens new window). | - Deletions - Duplications |
| Sawfish8 (opens new window) | Sawfish (opens new window) is a joint structural variant (SV) and copy number variant (CNV) caller for mapped HiFi sequencing reads. It discovers germline structural variants from local sequence assembly and jointly genotypes these variants across multiple samples. Sawfish replaces PBSV as the unified SV and CNV caller for PacBio HiFi data. | - Deletions - Duplications - Deletion-Insertions - Insertions - Breakends |
# Filtering Parameters Applied to SVs
Allele Fraction (AF) Filter: SVs with fractions lower than 0.2 are filtered out.
Pass Filter: SVs without the “PASS” flag assigned by their respective callers are filtered out.
Depth of Coverage (DP) Filter: SVs with fewer than 10 supporting reads are filtered out.
No Call Filter: SVs that have a 'no call' status in tandem repeat VCFS, ensuring that only fully determined genotypes are analyzed.
The Same Gene and Same Oriented Breakpoint (BND) Filter: Structural variants that involve the same gene and are oriented in the same direction are filtered out to reduce complexity and focus on more relevant genomic rearrangements.
Chromosome Filter: SVs that are not on chromosomes 1-22, X are filtered out.
Genomic Region Filter****: Variants overlapping with predefined blacklisted (Amemiya et al., 2019 (opens new window)) regions are filtered out. The complete list can be accessed here (opens new window).
**** Not applied to PacBio HiFi long-read WGS samples — the maximum SV size limit and the Genomic Region (blacklist) filter are skipped for this platform.
# Special Note on Repeat Finding
The repeat catalog focuses exclusively on tandem repeat regions known to cause diseases. For short-read and ONT long-read samples, we employ gnomAD's algorithm (opens new window) for detecting repeat unit motifs and then use ExpansionHunter on these de novo tandem repeat units to identify repeat sequences.
Occasionally, short-read sequencing technology falls short in accurately genotyping tandem repeats. In particular, tools like ExpansionHunter are not designed to genotype multiallelic repeats where different motifs might vary from each other. As a solution, we run ExpansionHunter for different motifs at the same locus to provide information for all repeat units present in the GnomAD. This approach helps capturing the variability and complexity of tandem repeats in genomic studies.
For PacBio HiFi long-read WGS samples, TRGT uses a normalized STRchive (opens new window) BED catalog (75 disease-associated loci (opens new window)) with IUPAC ambiguity code expansion in motif definitions. Each locus includes disease name, inheritance pattern, OMIM identifier, pathogenic and benign repeat count thresholds, and population frequency distributions from gnomAD.
# Targeted Variant Calling
Targeted variant calling is performed for PacBio HiFi long-read WGS samples.
# Targeted Variant callers used for Challenging Medically Relevant Genes
Paraphase (opens new window) (Chen et al., 2025 (opens new window)) runs on raw aligned BAM before phasing. Reads from each gene family are realigned to a single representative gene and then phased into individual haplotypes, enabling accurate copy number determination and variant calling across highly homologous segmental duplication regions. Paraphase supports 160 segmental duplication regions (opens new window); the following 14 medically relevant regions are integrated into the SEQ platform:
| Region | Genes | Small Variant Calling | Copy Number | Phasing |
|---|---|---|---|---|
| SMN1 (opens new window) | SMN1, SMN2 | Yes | Yes | — |
| PMS2 (opens new window) | PMS2, PMS2CL | Yes | Yes | — |
| STRC (opens new window) | STRC, STRCP1 | Yes | Yes | — |
| NCF1 (opens new window) | NCF1, NCF1B, NCF1C | Yes | Yes | — |
| IKBKG (opens new window) | IKBKG, IKBKGP1 | Yes | Yes | — |
| RCCX (opens new window) | CYP21A2, C4A, C4B, TNXB | Yes | Yes | Yes |
| NEB (opens new window) | NEB (exons 82–105 triplicate) | Yes | Yes (non-diploid) | Yes |
| GBA (opens new window) | GBA1, GBAP1 | Yes | Yes | Yes |
| CYP2D6 (opens new window) | CYP2D6, CYP2D7 (small variants: CYP2D6 only) | Yes | Yes | Yes |
| CYP11B1 (opens new window) | CYP11B1, CYP11B2 (small variants: CYP11B1 only) | Yes | Yes | Yes |
| HBA (opens new window) | HBA1, HBA2 (small variants: HBA2 only) | Yes | Yes | Yes |
| OPN1LW (opens new window) | OPN1LW, OPN1MW, OPN1MW2, OPN1MW3, TEX28 (small variants: OPN1LW only) | Yes | Yes | Yes |
| F8 (opens new window) | F8A1, F8A2, F8A3 | — | Yes | — |
| CFH (opens new window) | CFH, CFHR1, CFHR2, CFHR3, CFHR4 | — | Yes | — |
- Small Variant Calling: Paraphase produces haplotype-aware SNV/indel calls for these regions, assigning variants to the correct gene or its pseudogene/paralog copy. Within Paraphase target regions, Paraphase takes priority over other callers (e.g., DeepVariant). Other callers' variants that overlap with a Paraphase call are kept as co-called; the rest are filtered out with a
TargetedOverlapflag. Only Paraphase VCF metrics are displayed in the UI, and coverage is calculated from the Paraphase BAM. - Phasing: Individual haplotypes within the gene family are resolved and labeled, providing allele-level resolution for complex regions.
# Special Note: Non-Diploid Homozygous Effect
In non-diploid regions, variants detected in more than one but fewer than all available copies of a segmental duplication are marked with the  icon. For example, the NEB triplicate region (exons 82–105, Kiiski et al., 2016 (opens new window)) consists of three highly homologous copies per allele. A variant present on a subset of copies across both alleles may produce a homozygous functional effect — such as loss of function through a stop-gained variant — even though it is not carried by all copies of the region.
icon. For example, the NEB triplicate region (exons 82–105, Kiiski et al., 2016 (opens new window)) consists of three highly homologous copies per allele. A variant present on a subset of copies across both alleles may produce a homozygous functional effect — such as loss of function through a stop-gained variant — even though it is not carried by all copies of the region.
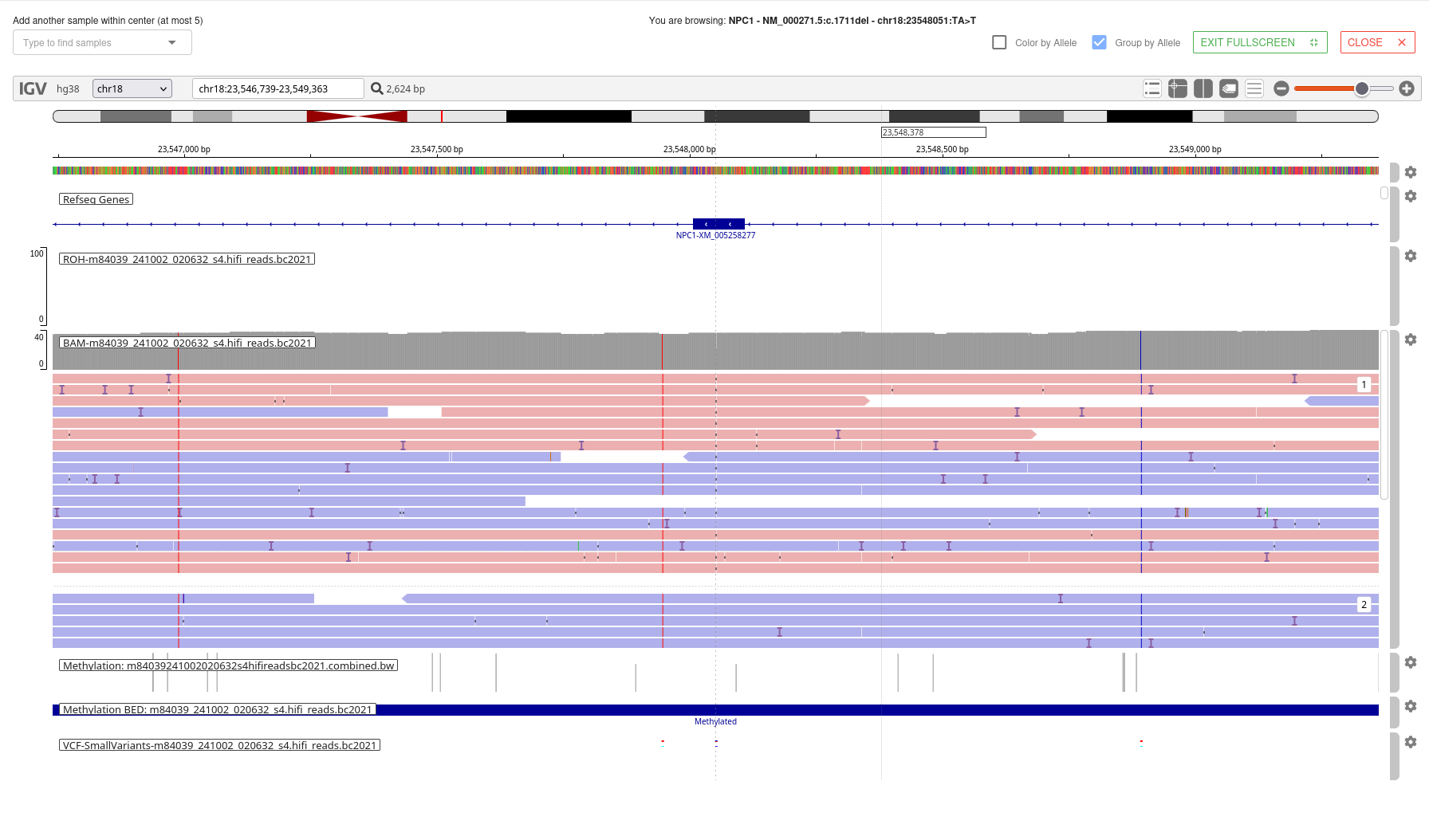
# Methylation Analysis
For PacBio HiFi long-read WGS samples, per-site CpG methylation scores are generated by pb-CpG-tools (opens new window) from aligned HiFi reads. Region-level methylation analysis is then performed by MethBat (opens new window), which produces two outputs per sample:
- Methylation Profile Summary — overall methylation statistics
- Methylation Regions — per-region methylation data as a BED track
CpG island regions are defined using the UCSC CpG Island reference (hg38). Methylation BED tracks can be visualized in the browser-embedded IGV, as seen below.
# Haplotype Phasing
For PacBio HiFi long-read WGS samples, haplotype phasing is performed by HiPhase (opens new window) (Holt et al., 2024 (opens new window)). HiPhase jointly phases small variants, structural variants, and tandem repeat variants using the aligned reads and variant calls, producing phased genotypes and haplotagged alignments. In segmental duplication regions, Paraphase provides full haplotype resolution for medically relevant gene families — see Targeted Variant Calling for details.
For ONT long-read WGS samples, haplotype phasing is performed using Sentieon's VariantPhaser and DNAscopeHP algorithms (Hu et al., 2025 (opens new window)). SNVs identified in the first pass are phased using long-read information, and variants in phased regions are then called from each haplotype separately.
# Special Note: Unresolved Homozygous Genotype
When a variant has been phased to one allele but could not be resolved in the other, the variant is marked with the  icon. The variant is reported as homozygous; however, the phasing information is incomplete as only one allele could be confidently assigned.
icon. The variant is reported as homozygous; however, the phasing information is incomplete as only one allele could be confidently assigned.
# Runs of Homozygosity (ROH) Analysis
The SEQ Platform uses PLINK v1.9 (Chang et al., 2015 (opens new window)) to perform Runs of Homozygosity (ROH) analysis, identifying contiguous stretches of homozygous genotypes within a sample's genome.
# ROH Detection Parameters
The following parameters are used for ROH detection:
| Parameter | Value | Description |
|---|---|---|
--homozyg-density | 50 | Maximum average distance between SNPs inside an ROH. Increasing this value relaxes the density requirement, enabling the identification of ROHs in regions with lower SNP density. |
--homozyg-gap | 1000 | Largest gap allowed between two consecutive homozygous SNPs. |
--homozyg-kb | 500 | Minimum length of a region to be reported as an ROH. |
--homozyg-snp | 50 | Minimum SNP count per ROH. |
--homozyg-window-het | 2 | Heterozygous calls tolerated in the sliding window. |
--homozyg-window-snp | 50 | SNPs required in the sliding window. |
# Sample Result
The sample dashboard summarizes key statistics from the ROH analysis, as shown in the example below.

- Total ROH Length: Sum of all identified ROH segment lengths.
- Average ROH Lengh: Mean length of all identified ROH segments.
- F-ROH: Proportion of the callable genome within ROH, estimating the level of inbreeding.
- ROH Variants: The number of variants located within all identified ROH segments.
- ROH Count Distribution by Length: The number of ROH segments grouped by ROH-length classes.
# ROH length classes
| Interval | Definition | Interpretation |
|---|---|---|
| < 1.5 Mb | Short ROH segments shorter than 1.5 Mb | Represents ancient or background homozygosity, often due to linkage disequilibrium. |
| 1.5 – 3 Mb | Intermediate ROH segments between 1.5 Mb and 3 Mb | Suggests distant parental relatedness (e.g., >3-5 generations ago) or endogamy. |
| 3 – 5 Mb | Long ROH segments between 3 Mb and 5 Mb | Suggests recent common ancestry (e.g., 2nd or 3rd cousins). |
| 5 Mb – 10 Mb | Very Long ROH segments between 5 Mb and 10 Mb | Provides moderate evidence for recent common ancestry (e.g., 2nd cousins). |
| ≥ 10 Mb | Ultra long ROH segments 10 Mb or longer | Provides strong evidence for recent common ancestry (e.g., 1st or 2nd cousins). |
*Adapted from Ceballos et al., 2018 (opens new window) and Gonzales et al., 2021 (opens new window)
# Small Variations
On the Small Variations page, variants located within a ROH segment are flagged with the  icon in the 'Genotype&Quality' column.
Hovering over the icon reveals the segment’s region, length, SNP count, and homozygosity ratio.
icon in the 'Genotype&Quality' column.
Hovering over the icon reveals the segment’s region, length, SNP count, and homozygosity ratio.
# ROH Filters
To filter for variants found within any ROH region, you can use the 'In ROH Region' toggle
( ).
).
The ROH Filters menu allows you to selectively display variants based on the characteristics of the ROH segments they are located in.

- Minimum/Maximum ROH Length (Mb): Filters for variants located in ROH segments that fall within the specified length range.
- Minimum SNP Count: Displays variants only from ROH segments containing at least the specified number of Single Nucleotide Polymorphisms (SNPs).
- Minimum Homozygosity Ratio: Restricts the view to variants in ROH segments that meet or exceed the defined ratio of homozygous variants.
# IGV integration
ROH intervals can be visualized as a BED track in the IGV.
# Genetic Sex Inference
The SEQ Platform includes an automated algorithm for chromosomal sex inference that assesses the heterozygosity ratio of single nucleotide polymorphisms (SNPs) across the non-pseudoautosomal regions (non-PAR) of the X chromosome, where patterns of variation differ between males (XY) and females (XX).
The sex inference result is displayed on the sample dashboard, as shown below.

To ensure reliable inference, a sample must meet predefined quality criteria for variant count and sequencing depth. Samples that do not meet these criteria have their analysis result reported as Not Available (NA).
For samples passing this quality control (QC) step, the algorithm applies validated thresholds to the X chromosome heterozygosity ratio to determine the inferred chromosomal sex:
- Female (XX): Assigned when the heterozygosity ratio exceeds the defined upper threshold, reflecting two X chromosomes and higher heterozygosity.
- Male (XY): Assigned when the heterozygosity ratio is below the defined lower threshold, consistent with the presence of a single X chromosome.
- Ambiguous: Assigned when the ratio falls within an intermediate range, where biological interpretation is less certain.
Benchmark data supporting the validation of this inference method are provided in the table below.
| Sex | Precision | Recall | F1 Score | Sample Count |
|---|---|---|---|---|
| Female | 0.988 | 0.981 | 0.984 | 576 |
| Male | 0.994 | 0.967 | 0.981 | 551 |
# Important Note: Limitations Regarding Chromosomal Anomalies
The sex inference algorithm is calibrated to distinguish between typical XX and XY chromosomal constitutions based on X chromosome heterozygosity. It is not designed or validated for the detection of sex chromosome aneuploidies (variations in the number of sex chromosomes) or other genetic anomalies.
Consequently, samples with certain chromosomal conditions may be misclassified by this method. For example:
- An individual with Turner Syndrome (X0) may be inferred as Male (XY) due to the presence of a single X chromosome.
- An individual with Klinefelter Syndrome (XXY) may be inferred as Female (XX) due to the heterozygosity pattern of two X chromosomes.
# gVCF Upload
# Run Selection
You can upload your samples to a new run by selecting “Create New Run” under “Run Name” and giving it a name on the “Name For New Run” field. You can also upload your samples to an existing run by selecting the previous run from the dropdown menu.
# Choose the Technology Type
Choose the next-generation sequencing machine associated with the samples. If you do not know which sequencing platform is used, you can select the "Unknown" option. Mixing different technologies in one run is not permitted.
# Choose the kit type
The SEQ platform has hundreds of different kits predefined in the system. A new kit can be defined with a set of target coordinates and a list of targeted genes. The Addition of a new kit typically takes one business day. For kit requests, please contact us through [email protected].
Every kit is associated with a standardized analysis version in SEQ. Probe-based kits, primer-based kits, Illumina & MGI technology, ION torrent technology, germline analysis, or somatic analyses all have a preset analysis version.
# Select the gVCF Files to be Uploaded
You can upload one small-variant gVCF file (for SNVs) per sample. Additionally, you may upload VCF files for CNVs, SVs, and STRs generated by DRAGEN or similar tools (See Supported Variant Callers) for the same sample, in any combination. Files are matched based on the sample information field, not the file names. Multisample VCFs are not supported.
# Submit your data
As the last step, you can upload your data by clicking the “Continue” button to start the upload process. After clicking "Continue", you will see the "Case Information" screen. Please refer to the "Genomize's Variant Prioritization" section for more information on entering the case information. You can then click the "Upload" button to see the number of analyses and the list of files matched for each analysis. Please be sure that both of these pieces of information are correct and hit "Approve" to start the upload process or "Cancel" to make changes.
When you start the upload, you will see the progress for each file. Transferred samples will immediately begin processing without waiting for the entire batch to finish uploading.
SEQ Platform's upload process is secure and performs a checksum to ensure the files are transferred correctly. Please do not close the browser tab or shut down your computer. Also, please ensure that your computer will not go into sleep/hibernation mode during the upload. Otherwise, the upload process will be aborted. Our upload process is resistant to intermittent loss of internet connection.
When the upload process is completed, you will be redirected to the corresponding Run's page, and your samples will be queued for analysis. Refresh the corresponding Run page to see the last status of the analysis.
# Supported Variant Callers
| Upload Type | Variant Types | Supported Callers | File Format (Extension) | Multisample Support |
|---|---|---|---|---|
| Small Variant | SNV INDEL | Dragen (v4.1, v4.2, v4.3, v4.4) | gVCF (.vcf.gz) (.g.vcf.gz) (.genome.vcf.gz) | Yes |
| Copy Number Variant | CNV | Dragen HifiCNV GATK-CNV Spectre ExomeDepth Sawfish | VCF (.vcf.gz) | No |
| Structural Variant | DEL (Deletion) DUP (Duplication) INV (Inversion) INS (Insertion) BND (Breakends)* CPX (Complex)** | Delly Dragen-SV Dragen-Targeted Callers (Coming Soon) Manta PBSV Sentieon Long Read SV Sniffles2 TIDDIT Sawfish cuteSV QDNAseq | VCF (.vcf.gz) | No |
| Short Tandem Repeat | STR*** | ExpansionHunter DRAGEN TRGT Straglr | VCF (.vcf.gz) | No |
| Targeted Caller Variant**** | DEL (Deletion) DUP (Duplication) | Dragen | JSON (.targeted.json) | No |
* BND (Breakends): 5'–3' fusion transcript events are supported, with potential formation of chimeric transcripts.
** CPX (Complex): Combination of structural variant types. Currently only DEL:INS variants are supported.
*** STR (Short Tandem Repeat): Only disease-associated STR variants in the gnomAD STR catalog are supported. For details, see the gnomAD STR catalog (opens new window).
**** Targeted Caller Variants: See Targeted Caller Variants section for the details.
# Unsupported Variant Callers and VCF Version Compatibility
Custom integration may be required to fully support unlisted or unsupported variant callers. Please contact support for assistance. Only VCF version 4.1 or newer is supported for Copy Number, Structural, and Short Tandem Repeat (STR) VCF files.
# Targeted Caller Variants
Small variants in gVCF files generated by Dragen targeted callers (opens new window) are supported. If the gVCF file already includes these variants, there is no need for a separate VCF file. However, if the variants are not included, they should be merged into the gVCF file prior to uploading. For assistance, please contact support.
Structural variants generated by Dragen targeted callers can be uploaded as an additional JSON file (opens new window). VCF files (opens new window) generated by Dragen targeted callers are currently not supported.
| Upload Type | Variant Types | Supported Callers | File Format (Extension) | Multisample Support |
|---|---|---|---|---|
| Structural Variant | DEL (Deletion) DUP (Duplication) | Dragen (Coming Soon) | VCF (.targeted.vcf.gz) | No |
| Targeted Caller Variant | DEL (Deletion) DUP (Duplication) | Dragen | JSON (.targeted.json) | No |
# Mitochondrial Variants
The Revised Cambridge Reference Sequence (rCRS, NC_012920.1) is used as the reference for the mitochondrial genome regardless of the genome version used in VCF generation, which is the recommended sequence for clinical use (McCormick et al., 2020 (opens new window)). If the VCF file contains variants called using the older Yoruban (YRI) mitochondrial reference genome, errors may result due to incompatibility with our annotation sources. Unsupported chrM variants should also be removed before upload to prevent genome compatibility issues. For assistance, or if issues arise, please contact support.
# Filtering Parameters Applied to Small Variants
No Call Filter: Small variants with a 'no call' status are excluded, ensuring that only fully determined genotypes are included in the analysis.
Chromosome Filter: Small variants that are not on chromosomes 1-22, X,M are filtered out.
# VCF Upload
# Run Selection
You can upload your samples to a new run by selecting “Create New Run” under “Run Name” and giving it a name on the “Name For New Run” field. You can also upload your samples to an existing run by selecting the previous run from the dropdown menu.
# Select the VCF Files to be Uploaded
You can upload VCF files for SNV, CNV, SV, and STR from DRAGEN or other similar tools (See Supported Variant Callers) for the same sample in any combination you choose. VCF files should have vcf.gzextension. Files are matched using the sample info field, not the file names. Multisample VCFs are not supported.
# Submit your data
As the last step, you can upload your data by clicking the “Continue” button to start the upload process. After clicking "Continue", you will see the "Case Information" screen. Please refer to the "Genomize's Variant Prioritization" section for more information on entering the case information. You can then click the "Upload" button to see the number of analyses and the list of files matched for each analysis. Please be sure that both of these pieces of information are correct and hit "Approve" to start the upload process or "Cancel" to make changes.
When you start the upload, you will see the progress for each file. Transferred samples will immediately begin processing without waiting for the entire batch to finish uploading.
SEQ Platform's upload process is secure and performs a checksum to ensure the files are transferred correctly. Please do not close the browser tab or shut down your computer. Also, please ensure that your computer will not go into sleep/hibernation mode during the upload. Otherwise, the upload process will be aborted. Our upload process is resistant to intermittent loss of internet connection.
When the upload process is completed, you will be redirected to the corresponding Run's page, and your samples will be queued for analysis. Refresh the corresponding Run page to see the last status of the analysis.
# Supported Variant Callers
| Upload Type | Variant Types | Supported Callers | File Format (Extension) | Multisample Support |
|---|---|---|---|---|
| Small Variant | SNV INDEL | DeepVariant Dragen Freebayes GATK - Haplotype Caller Ion Torrent Variant Caller Isaac Variant Caller Mutect2 Pivat Sentieon DNAscope, TNScope VarDict Clair3 Qiagen CLC | VCF (.vcf.gz) | No |
| Copy Number Variant | CNV | Dragen HifiCNV GATK-CNV Spectre ExomeDepth Sawfish | VCF (.vcf.gz) | No |
| Structural Variant | DEL (Deletion) DUP (Duplication) INV (Inversion) INS (Insertion) BND (Breakends)* CPX (Complex)** | Delly Dragen Manta PBSV Sentieon Long Read SV Sniffles2 TIDDIT Sawfish cuteSV QDNAseq | VCF (.vcf.gz) | No |
| Short Tandem Repeat | STR*** | ExpansionHunter DRAGEN TRGT Straglr | VCF (.vcf.gz) | No |
* BND (Breakends): 5'–3' fusion transcript events are supported, with potential formation of chimeric transcripts.
** CPX (Complex): Combination of structural variant types. Currently only DEL:INS variants are supported.
*** STR (Short Tandem Repeat): Only disease-associated STR variants in the gnomAD STR catalog are supported. For details, see the gnomAD STR catalog (opens new window).
# Unsupported Variant Callers and VCF Version Compatibility
Please note that using unlisted or unsupported variant callers may result in inaccurate VCF metrics. Unpredicted callers are categorized as "other," which may limit the capture of certain metrics. In some cases, custom integration may be required to support these callers fully. Only VCF version 4.1 or newer is supported for Copy Number, Structural and Short Tandem VCF files. If issues persist or critical data appears missing, please contact support for assistance.
# Mitochondrial Variants
The Revised Cambridge Reference Sequence (rCRS, NC_012920.1) is used as the reference for the mitochondrial genome regardless of the genome version used in VCF generation, which is the recommended sequence for clinical use (McCormick et al., 2020 (opens new window)). If the VCF file contains variants called using the older Yoruban (YRI) mitochondrial reference genome, errors may result due to incompatibility with our annotation sources. Unsupported chrM variants should also be removed before upload to prevent genome compatibility issues. For assistance, or if issues arise, please contact support.
# Genetic Sex Inference
The SEQ Platform includes an automated algorithm for chromosomal sex inference that assesses the heterozygosity ratio of single nucleotide polymorphisms (SNPs) across the non-pseudoautosomal regions (non-PAR) of the X chromosome, where patterns of variation differ between males (XY) and females (XX).
The sex inference result is displayed on the sample dashboard, as shown below.
To ensure reliable inference, a sample must meet predefined quality criteria for variant count and sequencing depth. Samples that do not meet these criteria have their analysis result reported as Not Available (NA).
For samples passing this quality control (QC) step, the algorithm applies validated thresholds to the X chromosome heterozygosity ratio to determine the inferred chromosomal sex:
- Female (XX): Assigned when the heterozygosity ratio exceeds the defined upper threshold, reflecting two X chromosomes and higher heterozygosity.
- Male (XY): Assigned when the heterozygosity ratio is below the defined lower threshold, consistent with the presence of a single X chromosome.
- Ambiguous: Assigned when the ratio falls within an intermediate range, where biological interpretation is less certain.
Benchmark data supporting the validation of this inference method are provided in the table below.
| Sex | Precision | Recall | F1 Score | Sample Count |
|---|---|---|---|---|
| Female | 0.988 | 0.981 | 0.984 | 576 |
| Male | 0.994 | 0.967 | 0.981 | 551 |
# Important Note: Limitations Regarding Chromosomal Anomalies
The sex inference algorithm is calibrated to distinguish between typical XX and XY chromosomal constitutions based on X chromosome heterozygosity. It is not designed or validated for the detection of sex chromosome aneuploidies (variations in the number of sex chromosomes) or other genetic anomalies.
Consequently, samples with certain chromosomal conditions may be misclassified by this method. For example:
- An individual with Turner Syndrome (X0) may be inferred as Male (XY) due to the presence of a single X chromosome.
- An individual with Klinefelter Syndrome (XXY) may be inferred as Female (XX) due to the heterozygosity pattern of two X chromosomes.
# Filtering Parameters Applied to Small Variants
No Call Filter: Small variants with a 'no call' status are excluded, ensuring that only fully determined genotypes are included in the analysis.
Chromosome Filter: Small variants that are not on chromosomes 1-22, X,M are filtered out.
# Cloud Browser
To use the cloud browser, select or create your run, select the sequencing platform and the kit by following the directions above. After the kit selection, you will see the option to select either your “COMPUTER” or the “CLOUD BROWSER” as the data source.
When you select the "CLOUD BROWSER" option, click the PLUS (➕) button to open the cloud browser interface. Using the cloud browser, you can choose the files with which you want to start the analysis and click “DONE”. The rest of the process is the same as described above. Please note that there is a 3-minute duration between each cloud upload process, and files will be removed from your cloud account upon starting the analysis.