# Cohort CNV Analysis
IMPORTANT NOTE : Please refer to our technical paper about the clinical validation of our CNV analysis explaining why we decided to use the GATK CNV pipeline. This clinical validation study was executed by using more than 250 data points from MLPA verifications of a deeply-sequenced cohort of 96 samples. At 600x depth, we discovered that our CNV analysis pipeline provides 95.46% sensitivity and 99.72% specificity. The samples must be prepared on the same day and sequenced in the same run. You can see the effect of depth and the cohort size on sensitivity and specificity in our technical paper (opens new window) .
Current CNV analysis version is adapted from GATK CNV algorithm[1].
# Special Note on Detection of Deletions
To increase the sensitivity of deletion detection in exome kits and gene panels targeting over 100 genes, the split-read method is employed using Delly (opens new window) as an additional variant caller.
# Filtering Parameters Applied to CNVs Detected by the Split-Read Method (Delly)
Pass Filter: Deletions without the “PASS” flag are filtered out.
Split-Read Depth of Coverage Filter: Deletions supported by fewer than 5 split-reads are filtered out.
Imprecise Deletion Filter: Deletions without precise breakpoint positions are filtered out.
Length Filter: Deletions longer than 100 kbp (100,000 base pairs) are filtered out.
# Start the Analysis
A minimum of eight (8) samples in the same run is required to start a CNV analysis, and a single sample can only be included in one CNV analysis. For optimal results, individual samples should be from the same sequencing run, unrelated to each other, and of the same sex. Including samples of different sexes in the analysis may cause unreliable results for X and Y chromosomes.
To start the GATK CNV Analysis, you can go to the "Runs" page and click on the "CNV Analysis" button in the "CNV" column. Alternatively, you can go to the sample dashboard of one of your samples and click on the "Start New CNV Analysis" option in the menu on the CNV card.
You can also start a CNV analysis by going to the sample dashboard, clicking the "..." icon top-right, and selecting "New CNV analysis.”
You can include all available samples in the run. You can also select the samples yourself. Then, you can choose the samples for the sample group. Next, you can select the kit that you want to start the analysis at this step in case there is more than one kit in the run. You cannot mix and match samples prepared with different kits.
You can also adjust the "Sample Incidence Threshold” at this step. The default value is half of the sample count. Any CNV that occurs in more than half of the samples (or the adjusted threshold value) will be filtered out. You can use the toggle to switch the threshold off to list every CNV detected in your sample regardles of the occurence within the analysis cohort.
When you are ready, click on "Start Analysis" button.
# Results
To see the CNV results, open a sample and click on the CNV tab on the top right.
Gene Info: In this column you will se the gene(s) affected by the reported CNV. If a single gene is affected,
you will also see the information about the affected region
My Verdict: You can change the predicted pathogenicity of the reported CNV in this column. This change will be associated
with the reported CNV. You will also see your pathogenicity selection for this CNV in your other samples.
ACMG: Here you will see the pathogenicity calculated according to the ACMG & Clingen guideline for pathogenicity
calculation for CNVs (opens new window).
Copy Number: The copy number of this CNV in your current sample is listed here.
Occurence: This column shows the distribution of the reported CNV among your analysis cohort.
Del shows the number of samples
with a deletion in this region.
Dup shows number of samples with duplications.
Mixed shows the number of samples with a combination of del, dup or wt in this region.
Wt shows the number of samples with no detected CNVs in this region.
Dosage: This field shows the haploinsufficiency (HI) and triplosensitivity (TS) scores from ClinGen, if available. You can click on these
scores to access the ClinGen's page for the gene's dosage sensitivity page. Please refer to ClinGen's
publication (opens new window) for details.
Constraint: Here, you will find pLI and LOEUF scores for the gene(s) from GnomAD database, if available. For details on
these values and how they are calculated, please refer to GnomAD's official help page (opens new window).
Related Diseases: In this column, you can see diseases associated with the gene in question in curated databases
as well as the mode of inheritance of the disease.
HPOs: Here, the number of matched HPO entries to the clinical information you provided in the sample upload or the
phenotype filter you applied using the filters.
You can get detailed information about the CNV using the “Detailed information” field at the bottom of the page.
# CNV Occurrence within the Run (Cohort)
The "Occurrence within Run" column shows the number of samples in the cohort that have a deletion, duplication, wildtype or mixed state in the CNV region.
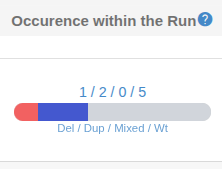
In the details panel, the "Occurrence" tab displays a graph of Copy Number Variations (CNVs) across the analysis cohort, broken down by sample, and also includes a list of the wildtype samples.

# CNV Overlap with Other Samples (CNV Database)
The "Overlapping CNVs" column shows the number of samples with overlapping CNV calls separately for deletions and duplications.
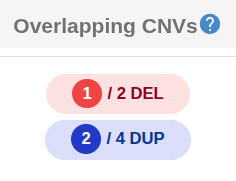
The following filters are applied in the database:
- Minimum Overlap Percentage (Default: 25%): This value is calculated by dividing the length of the intersection by the length of the union of the two CNV intervals.
- Containment: A CNV record from database is counted as a hit if it is fully contained within the query CNV, regardless of the overlap percentage.
- Cohort (Run) filters:: Set the minimum run sample size (Default: 20) and the maximum sample occurrence (Default: 3), which is calculated as the sum of deletions, duplications, and mixed types.
These settings can be changed in Site Settings (for all future sessions) or in the Settings section under the CNV tab (for the current session only).
Combining these filters is an effective way to remove a significant number of background CNV calls, helping you focus on those that are biologically relevant.
# CNV Database – Detailed Sample View
You can click over the "DEL" or "DUP" buttons located in the "Overlapping CNVs" column to see the specific samples with overlapping CNV calls. This detailed view allows you to review relevant sample information and apply filters based on overlap percentage, cohort criteria, and variant length.
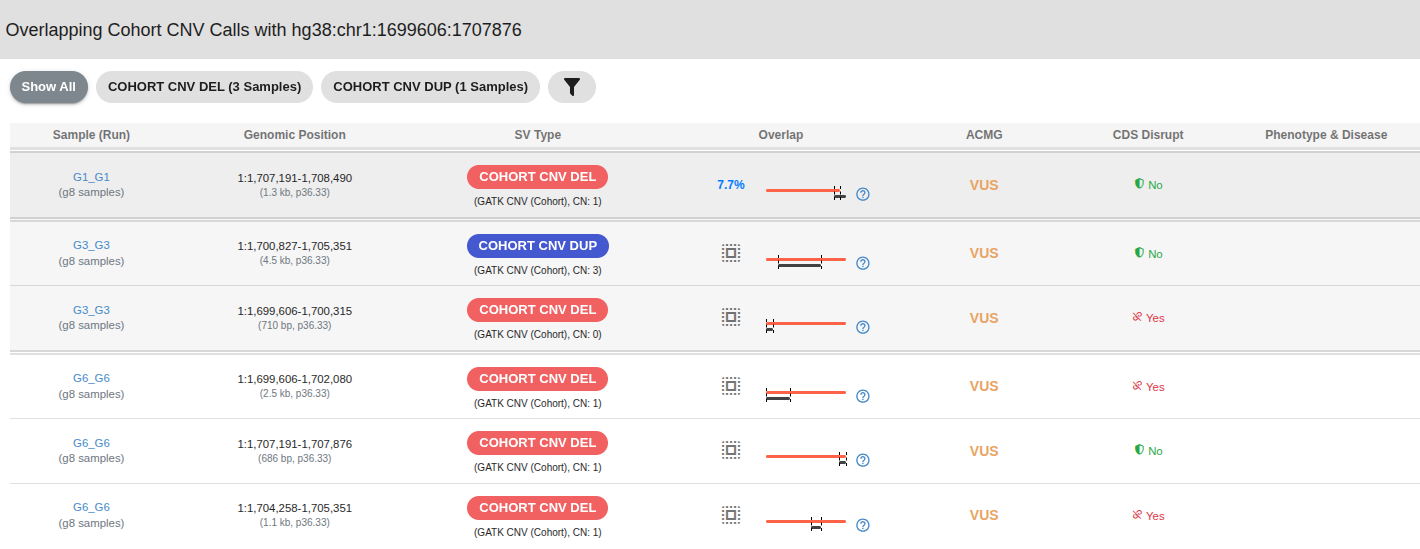
Table shows the following information:
- Sample and Run Name
- Genomic Position
- CNV/SV Type
- Overlap: Shows the overlap percentage or indicates if the query CNV contains the database CNV.
- ACMG Pathogenicity
- Phenotype & Disease: Phenotype/Disease information entered by the user
- CDS Disruption: Indicates if the CNV disrupts the protein-coding sequence of a transcript. This is determined by whether the CNV interrupts the Open Reading Frame (ORF) of any transcript within that region.
# Small Variant Page: Viewing Overlapping CNVs
On the small variant page, you can hover over the "DEL" or "DUP" buttons in the "Genotype & Quality" column to view CNVs from the same sample that overlap with the transcript where the small variant is located.
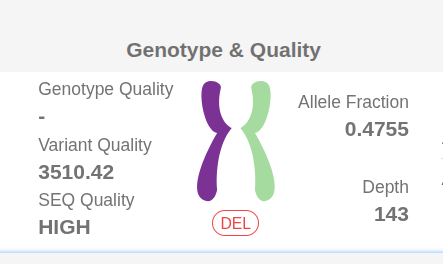
A table will appear displaying the overlapping CNVs and providing key annotation details, including:
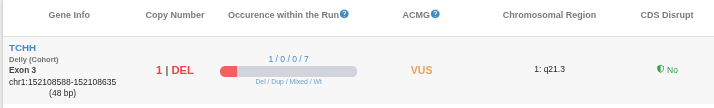
- Genomic Coordinates and Gene Info
- Copy Number
- Occurrence within the Run
- ACMG Pathogenicity
- Chromosomal Region
- CDS Disruption: Indicates if the CNV disrupts the protein-coding sequence of a transcript. This is determined by whether the CNV interrupts the Open Reading Frame (ORF) of any transcript within that region.
# Copy Number Variant Annotation
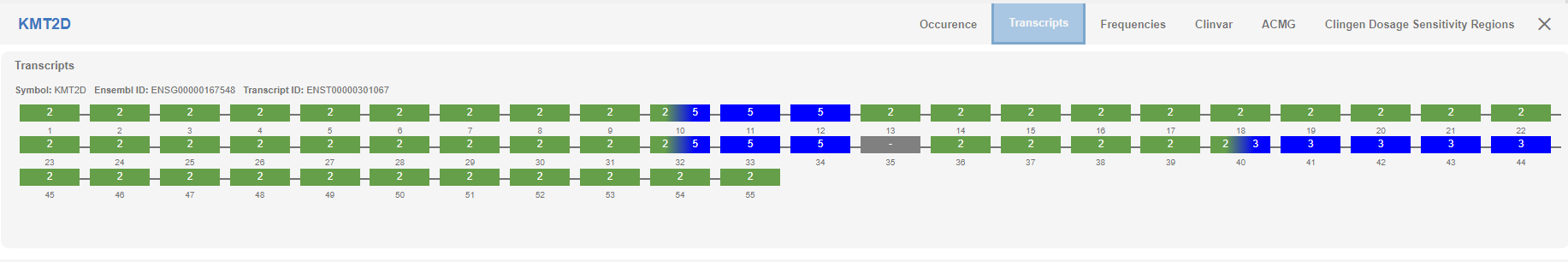
# Transcripts
In this tab, you will find the graphical representation of the transcript(s) and the copy number states of the gene(s) affected by the CNV.
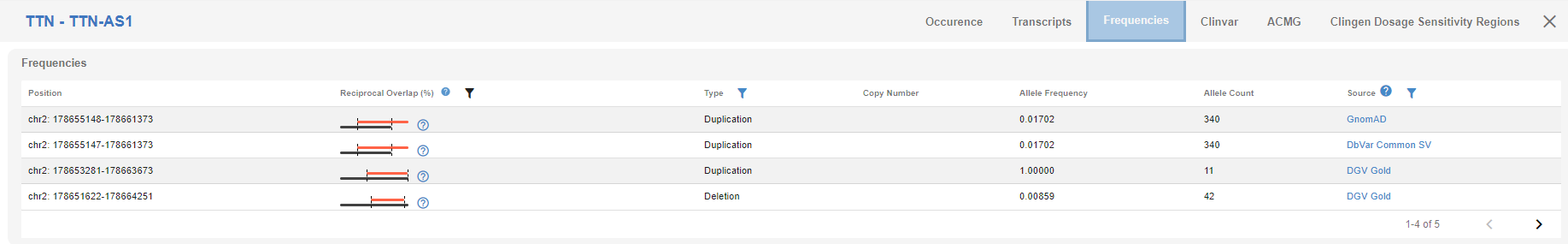
# Frequencies
Here, you will see the frequency and reciprocal overlap of the CNV in various databases. You can use the quick links on the right to access the entries in the designated database. You can also filter the entries using the "funnel" icon on different columns.
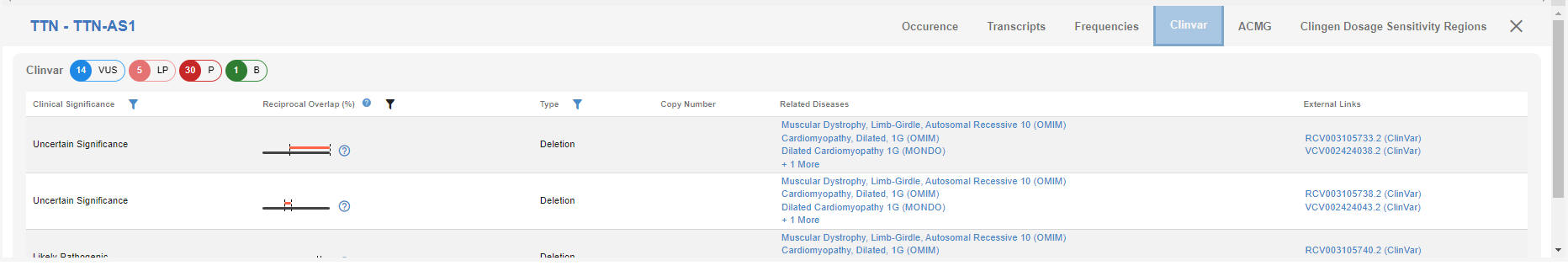
# ClinVar
ClinVar entries for the CNVs that intersect with the detected CNV are shown in this tab. You can use the links in the "Related Diseases" column to access entries in their respective databases. "RV” and “CV” entries in the ClinVar database can be accessed using the links in the "External Links" column. You can also see the reciprocal overlap of the CNV with the ClinVar entries. You can use the "funnel" icon in different columns to filter the results from the ClinVar database.
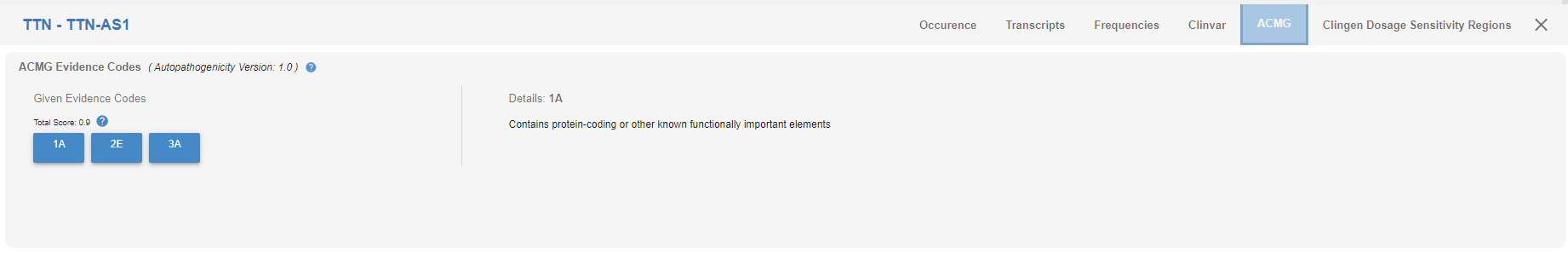
# ACMG
ACMG evidence codes assigned to the CNV and their explanations can be found here.
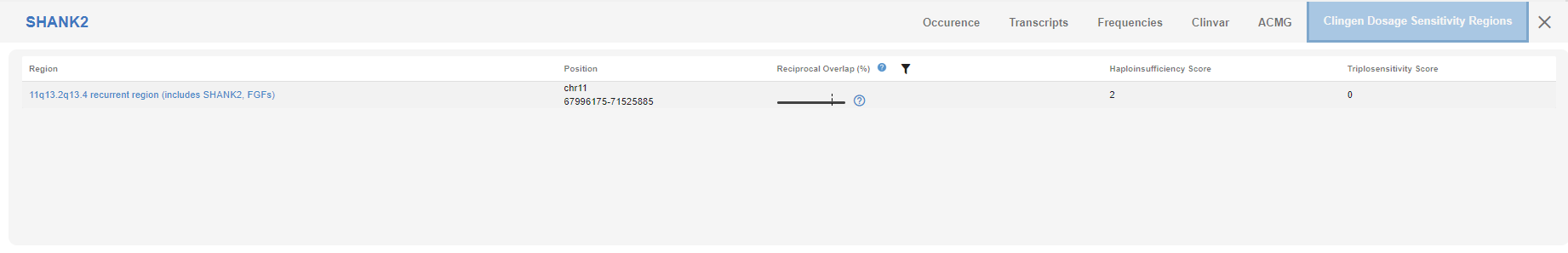
# ClinGen Dosage Sensitivity Region
If there are any entries available in the ClinGen database for this region, it will be shown in this tab.
DISCLAIMER: CNV analysis is for research use only. Genomize does not accept any responsibility for liabilities that may arise from unintended use such as, but not limited to, diagnosis.
[1] “(How to) Call common and rare germline copy number variants – GATK.” [Online]. Available at: https://gatk.broadinstitute.org/hc/en-us/articles/360035531152--How-to-Call-common-and-rare-germline-copy-number-variants (opens new window) [Accessed: 24-July-2023].