
# Structural Variant Detection and Visualization in WGS Samples
SEQ Platform can detect and report various structural variants listed below. For single samples, maximum SV size is limited to 100,000 bps.
For cohort CNV analysis in WGS samples, please refer to Copy Number Variations section
# Supported Structural Variants
| SV-Group | Abbreviation | Supporting callers |
|---|---|---|
| Deletion | DEL | Manta, Tiddit, Delly, PBSV, HifiCNV, DragenCNV, Paraphase* |
| Duplication | DUP | Manta, Tiddit, Delly, PBSV, HifiCNV, DragenCNV, Paraphase* |
| Insertion | INS | Manta, Delly, PBSV |
| Inversion | INV | Tiddit, Delly, PBSV |
| Breakend (Unresolved)** | BND | Manta, Tiddit, Delly, PBSV |
| Short Tandem Repeat | STR | ExpansionHunter, TRGT |
| Complex*** | CPX | Manta, Tiddit, Delly |
* Coming Soon
** Structural variants that cannot be classified into any other type are listed as BND
*** If more than one type of SV is detected in combination, they are classified as CPX variant. ex: DEL:INS, DUP:INV, etc. Currently only DEL:INS variants are supported.
# Variant callers used for SV detection
| Tool | Algorithm | Supported SV-types |
|---|---|---|
| Manta1 (opens new window) | Manta (opens new window) divides the SV and indel discovery process into two primary steps: 1. Scanning the genome to find SV associated regions. 2. Analysis, scoring and output of SVs found in these regions. | - Deletions - Duplications - Deletion-Insertions - Insertions - Breakends |
| Delly2 (opens new window) | DELLY (opens new window), short-range and long-range paired-end libraries are analyzed for discordantly mapped read pairs. Paired-end predicted structural variants are then refined using split-reads and reported at single-nucleotide breakpoint resolution. In addition to general parameters applied to SVs, insert size cutoff for split reads ≥ 15 bps, minimum paired-end MAPQ ≥ 20 filters are used for DELLY. | - Deletions - Duplications - Deletion-Insertions - Insertions - Inversions - Breakends |
| Tiddit3 (opens new window) | TIDDIT (opens new window), detects structural variants by examining sequences for discordant pairs, split reads, and supplementary alignments, which must exceed a specified quality threshold. It uses a clustering method similar to DBSCAN, where a cluster forms if sufficient signals are within a designated distance. Clusters lacking enough signals are discarded; otherwise, they are included in the output regardless of other quality filters. | - Deletions - Duplications - Inversions - Breakends |
| ExpansionHunter4 (opens new window) | ExpansionHunter (opens new window) is a tool designed for targeted genotyping of short tandem repeats (STRs) and flanking variants. It operates by analyzing BAM files to find reads that either span, flank, or are fully contained within each targeted repeat. This precise approach allows for effective characterization of these genomic elements, tailored specifically to identify and quantify repeat variations. | Short Tandem Repeats |
| PBSV5 (opens new window) | PBSV (opens new window) is a suite of tools to call and analyze structural variants in diploid genomes from PacBio single molecule real-time sequencing (SMRT) reads. | - Deletions - Duplications - Insertions - Inversions - Breakends |
| HifiCNV6 (opens new window) | HifiCNV (opens new window) is a cutting-edge tool specifically designed for calling copy number variants (CNVs) using high-fidelity (HiFi) sequencing reads. It offers optimized segmentation and calling for germline whole genome sequencing (WGS) using HiFi reads, ensuring accurate results. The tool automatically estimates and corrects GC-bias, which enhances the reliability of the data. | - Deletions - Duplications |
| TRGT7 (opens new window) | TRGT (opens new window) is a tool for targeted genotyping of tandem repeats from PacBio HiFi data. In addition to the basic size genotyping, TRGT profiles sequence composition, mosaicism, and CpG methylation of each analyzed repeat and visualization of reads overlapping the repeats. | Short Tandem Repeats |
| DragenCNV8 (opens new window) | DragenCNV identifies CNV events using next-generation sequencing (NGS) data, applicable to both whole-genome (WGS) and whole-exome sequencing (WES) for germline analysis. The pipeline includes modules for counting reads, correcting biases, normalizing ploidy levels, segmenting the genome to detect breakpoints, and calling or genotyping the variants. It supports two normalization modes tailored to different applications, ensuring optimal accuracy and speed. Additionally, a panel of normals (PoN) can be used for more refined normalization when available, enhancing the accuracy of CNV detection. | - Deletions - Duplications |
| Paraphase (Coming Soon)9 (opens new window) | Paraphase (opens new window) is a Python tool that takes HiFi aligned BAMs as input (whole-genome or enrichment), phases haplotypes for genes of the same family, determines copy numbers and makes phased variant calls. Paraphase supports 160 segmental duplication regions (opens new window). | - Deletions - Duplications |
# Filtering Parameters Applied to SVs
Allele Fraction (AF) Filter: SVs with fractions lower than 0.2 are filtered out.
Pass Filter: SVs without the “PASS” flag assigned by their respective callers are filtered out.
Depth of Coverage (DP) Filter: SVs with fewer than 10 supporting reads are filtered out.
No Call Filter: SVs that have a 'no call' status in tandem repeat VCFS, ensuring that only fully determined genotypes are analyzed.
The Same Gene and Same Oriented Breakpoint (BND) Filter: Structural variants that involve the same gene and are oriented in the same direction are filtered out to reduce complexity and focus on more relevant genomic rearrangements.
Chromosome Filter: SVs that are not on chromosomes 1-22, X are filtered out.
# Special Note on Repeat Finding
The repeat catalog focuses exclusively on tandem repeat regions known to cause diseases. We employ gnomAD's algorithm for detecting repeat unit motifs and then use ExpansionHunter on these de novo tandem repeat units to identify repeat sequences.
Occasionally, short-read sequencing technology falls short in accurately genotyping tandem repeats. In particular, tools like ExpansionHunter are not designed to genotype multiallelic repeats where different motifs might vary from each other. As a solution, we run ExpansionHunter for different motifs at the same locus to provide information for all repeat units present in the GnomAD. This approach helps capturing the variability and complexity of tandem repeats in genomic studies.
# ACMG Classification
Due to currently available guidelines, ACMG classification is only available for DEL and DUP variants.
# Structural Variants Page
When the analysis of your WGS sample is completed, you can click on the "Structural Variants" tab to view and filter detected structural variants in your sample.
Similar to small variants tab, detected structural variants are listed at the top, and the details are shown at the bottom of the page when you click on a variant.
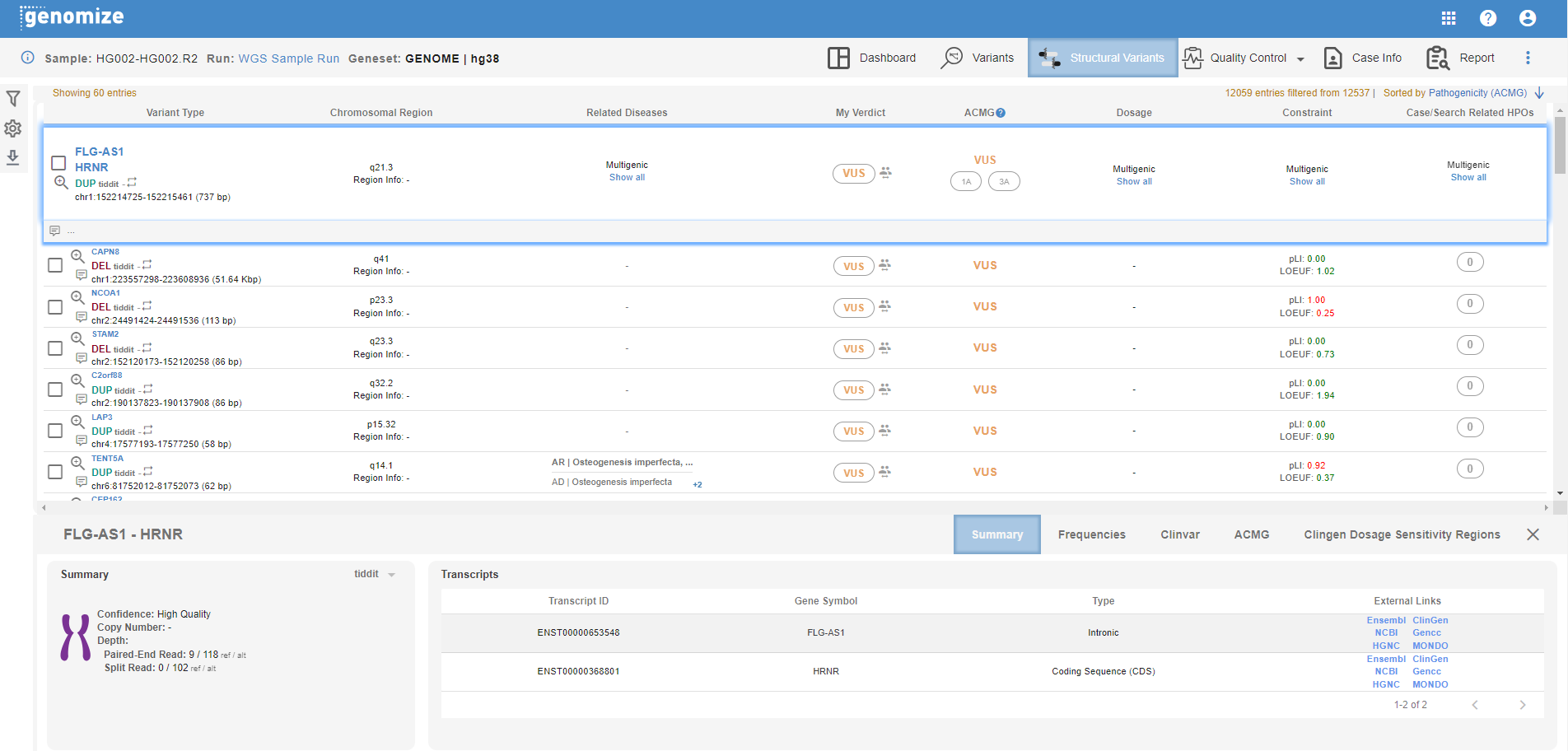
The columns in the list are:
- Variant Type: This field shows the effected gene(s), SNV type, the variant caller that detected the variant, and the chromosomal coordinates and the size of the variant. You can use the report checkbox to include the variant in your report, and use the IGV button to visualize the variant in desktop or web browser IGV depending on your IGV setting.
- Chrosomal Region: The corresponding cytoband is shown here.
- Related Diseases: Disease(s) associated with the affected genes in various databases are shown here.
- My Verdict: You can change the pathogenecity of the variant in this field. The default value is the ACMG pathogenecity calculated by the SEQ Platform. You can also see the statistics for pathogenecity change(s) by other centers by mousing over the people icon.
- ACMG: This field shows the ACMG pathogenecity calculated by the SEQ Platform using the relevant guideline(s). Currently, this information is only available for DEL and DUP SV types.
- Dosage: Haplo insufficiency (HI) and triplosensitivity (TS) information from ClinGen are shown here. For multigenic SVs, click on "show all" to see the list of genes and their corresponding dosage information.
- Constraint: pLI and LOEUF values from GnomAD are shown here. For multigenic SVs, click on "show all" to see the list of genes and their corresponding constraint information.
- Case/Search Related HPOs: Number of matching HPOs is shown here. Click on the variant line to expand the information and see the list of matching HPOs.
At the bottom of your screen you will see the details section. Available tabs and information change based on the SV type:
- Summary: Available for DEL, DUP, STR, and BND. Various information such as zygosity, read counts, supporting read information, etc. are shown here. Available information may change based on SV type.
- Frequencies: Available for DEL, DUP, BND, INS, CPX, and INV. Frequencies in various databases are shown here, if available.
- ClinVar: Available for DEL, DUP, INS, CPX, and INV. List of ClinVar entries and corresponding links are shown here.
- ACMG: Available for DEL and DUP. ACMG pathogenicity calculation, applied evidence codes, and aditional information is shown here.
- ClinGen Dosage Sensitivity Regions: Available for DEL, DUP, BND, INS, CPX, and INV. Additional curation information from ClinGen for the affected genomic regions is shown here, if available.
- Repeat Number Distribution: Available for STR only. The repeat number distribution of the repeat unit is shown here with an interactive graph. You can visualize the distribution for the global data or for available ethnicities.