
# Small Variations
# Calling
SEQ has standard analysis versions pre-setup for every kit defined in the system. Data processing and variant calling are handled differently based on the sample type, sequencing platform, and selected analysis pipeline.
# Analysis versions for long-read WGS
| Name | Explanation | Alignment/ variant calling | CMRG Support* | SV Calling | Phasing | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|---|
| Sentieon minimap2-DNAScope-SV Calling-Phasing-germline | Optimized for ONT long-read WGS samples | Sentieon minimap2 / DNAScope | No | Sention LongreadSV | VariantPhaser | hg38 | Oxford Nanopore |
| PacBio pbmm2-DeepVariant-SV calling-Phasing-germline | Optimized for Pacbio long-read WGS samples | Pacbio pbmm2 / Deepvariant | **Paraphase (Coming soon) | PBSV, HifiCNV, TRGT | HiPhase | hg38 | Pacbio |
* CMRG: Challenging Medically Relevant Genes (Wagner et al., 2022 (opens new window))
** Paraphase (Coming Soon): PacBio's recommended tool for detecting segmental duplication regions and medically relevant genes, such as SMN1/SMN2 and HBA1/HBA2 (Chen et al., 2024 (opens new window)). Refer to the table here (opens new window) for a complete list of regions.
# Analysis versions for short-read WGS
| Name | Explanation | Alignment/ variant calling | BAM processing | SV Calling | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|
| Sentieon BWA-DNAScope-SV Calling-germline | Optimized for WGS samples prepared with a PCR enrichment step | Sentieon BWA / DNAScope | MarkDuplicate | Delly, Manta, Tiddit, ExpansionHunter | hg19, hg38 | Illumina, MGI |
| Sentieon BWA (PCRfree)-DNAScope-SV Calling-germline | Optimized for WGS samples prepared without a PCR enrichment step | Sentieon BWA / DNAScope | MarkDuplicate | Delly, Manta, Tiddit, ExpansionHunter | hg19, hg38 | Illumina, MGI |
| BWA-Freebayes-SV Calling-germline | Optimized for WGS samples | BWA / Freebayes | PCR Dedup + Indel Realignment | Delly, Manta, Tiddit, ExpansionHunter | hg19, hg38 | Illumina, MGI |
| BWA-GATK-SV Calling-germline | Optimized for WGS samples | BWA / GATK | PCR Dedup + Indel Realignment | Delly, Manta, Tiddit, ExpansionHunter | hg19, hg38 | Illumina, MGI |
# Analysis versions for capture based targeted panels, including WES
| Name | Explanation | Alignment/ variant calling** | BAM processing | CNV Calling (Cohort Mode) | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|
| Sentieon BWA-DNAScope- germline | Optimized for capture-based germline kits. | Sentieon BWA / DNAScope | MarkDuplicate | GATK-CNV + delly* | hg19, hg38 | Illumina, MGI |
| BWA-Freebayes-PCR dedup - germline | Optimized for capture-based germline kits. | BWA / Freebayes | PCR Dedup | GATK-CNV + delly* | hg19, hg38 | Illumina, MGI |
| BWA-Freebayes-PCR dedup-Indel Realignment - germline | Optimized for capture-based germline kits. Default analysis for most kits. | BWA / Freebayes | PCR Dedup + Indel Realignment | GATK-CNV + delly* | hg19, hg38 | Illumina, MGI |
| BWA-GATK-PCR dedup-Indel Realignment - germline | Optimized for capture-based germline kits. Uses a GATK variant caller. | BWA / GATK | PCR Dedup + Indel Realignment | GATK-CNV + delly* | hg19, hg38 | Illumina, MGI |
| BWA-Freebayes High Sensitivity-PCR dedup-Indel Realignment - germline | Optimized for capture-based germline kits to call variants with a low fraction (<20%). | BWA / Freebayes | PCR Dedup + Indel Realignment | GATK-CNV + delly* | hg19, hg38 | Illumina, MGI |
* Delly is utulized only for panels with more than 100 genes.
** Mitochondrial analysis is performed following GATK best practices, and gnomAD filters are applied (for details, see Mitochondrial Calling).
# Analysis versions for amplicon based panels
| Name | Explanation | Alignment/ variant calling | BAM processing | Primer Trimming | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|
| BWA-Freebayes-BamKeser-Indel Realignment - germline | Optimized for amplicon-based germline kits. | BWA / Freebayes | Indel Realignment | BamKeser | hg19, hg38 | Illumina, MGI |
| BWA-Freebayes-BamKeser - germline | Optimized for amplicon-based germline kits. Does not perform indel realignment step. | BWA / Freebayes | NA | BamKeser | hg19, hg38 | Illumina, MGI |
| CVD Specific-BWA-Bowtie2-Freebayes-Bamkeser - germline | Optimized for amplicon-based CVD kits. | BWA + Bowtie2 / Freebayes | NA | BamKeser | hg19 | Illumina, MGI |
| Thalassemia Specific-BWA-Freebayes-Bamkeser - germline | Optimized for amplicon-based thalassemia kits. | BWA / Freebayes | NA | BamKeser | hg19 | Illumina, MGI |
| CAH Specific v2-BWA-Freebayes-Bamkeser - germline | Optimized for amplicon-based CAH kits. | BWA / Freebayes | NA | BamKeser | hg19 | Illumina, MGI |
**BamKeser is our in-house designed and precisely working primer trimming tool.
# Analyis versions for IonTorrent uBAM samples
| Name | Explanation | Alignment/ variant calling | BAM processing | Primer Trimming | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|
| Torrent Suite 5.8 - No Trimming - Default Parameters v2 - germline | Optimized for IonTorrent samples. Does not perform primer trimming. | Torrent Suite 5.8 | NA | NA | hg19 | IonTorrent |
| Torrent Suite 5.8 - No Trimming - BRCA specific - germline | Optimized for IonTorrent BRCA samples. | Torrent Suite 5.8 | NA | NA | hg19 | IonTorrent |
| Torrent Suite 5.8 - No Trimming - CFTR specific - germline | Optimized for IonTorrent CFTR samples. | Torrent Suite 5.8 | NA | NA | hg19 | IonTorrent |
**BamKeser is our in-house designed and precisely working primer trimming tool.
After the variant calling, the Genomize-SEQ processes the resulting VCF file to form a Genomize standard VCF file which can be downloaded through the platform. The Genomize standard VCF line will have gstd=1 in the info field. Standardization of the VCF file includes the following important steps
- Minimal variant representation: Some callers produce redundant bases at the left-hand or right-hand side of either alternative or reference allele. This redundancy has to be removed to obtain the correct annotation of variants in the subsequent steps.
# Mitochondrial Calling
The mitochondrial variant calling pipeline follows GATK best practices (opens new window), using the revised Cambridge Reference Sequence (NC_012920.1) as the reference mitochondrial genome and the Mutect2 as variant caller.
Filtering and genotype assignment for mitochondrial variants are performed in line with the recommendations from gnomAD (opens new window) . Briefly, SNVs with Variant Allele Frequency (VAF) below 0.01 were removed. Variants with VAF equal to or higher than 0.95 are classified as homoplasmic. Variants with VAF lower than 0.95 are classified as heteroplasmic. Any variants in previously reported artifact-prone sites (positions 301, 302, 310, 316, 3107, and 16182) are ignored.
In targeted panel analyses (including WES), mitochondrial variant detection is only available for panels with distinct chrM targets in their bed files.
In WGS analyses, mitochondrial variant detection uses the same variant callers and filtering steps as those applied to other chromosomal regions. If your WGS sample preparation is optimized for mitochondrial DNA isolation and enrichment, please contact support for further assistance.
# Annotation
Genomize standard VCFs are annotated by using the VEP (opens new window) tool from NCBI which is a trusted and constantly developed open-source annotation tool. Computational prediction algorithms are provided by VEP including sift, polyphen, mutationtaster, revel, metalR, and DANN.
In addition, the variants are annotated with the dbSNP database and the ClinVar database.
# Extended Annotation
Genomize offers the "Extended Annotation Service" to all SEQ Platform users as a part of its basic services. With the "Extended Annotation Service", our users can see the variants in the non-reference Ensembl and the RefSeq transcripts in the variant list if these variants increase the pathogenicity. With this functionality, it is now even less likely to "miss" a variant using the SEQ Platform.
The variants listed thanks to the "Extended Annotation Service" are marked with the  icon on the variant list.
icon on the variant list.
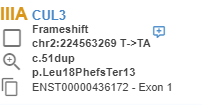
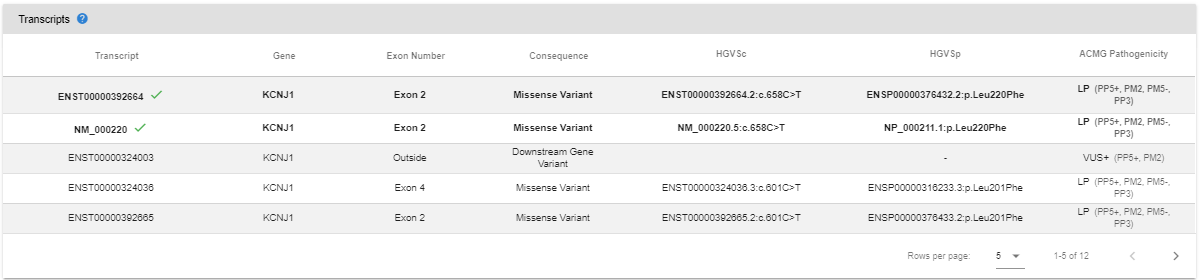
You can also see the ACMG pathogenicity predictions for all known transcripts on the variant page.
# Visualization
# Variants Tab
The information related to a variant can be visualized in the "Variants" tab.
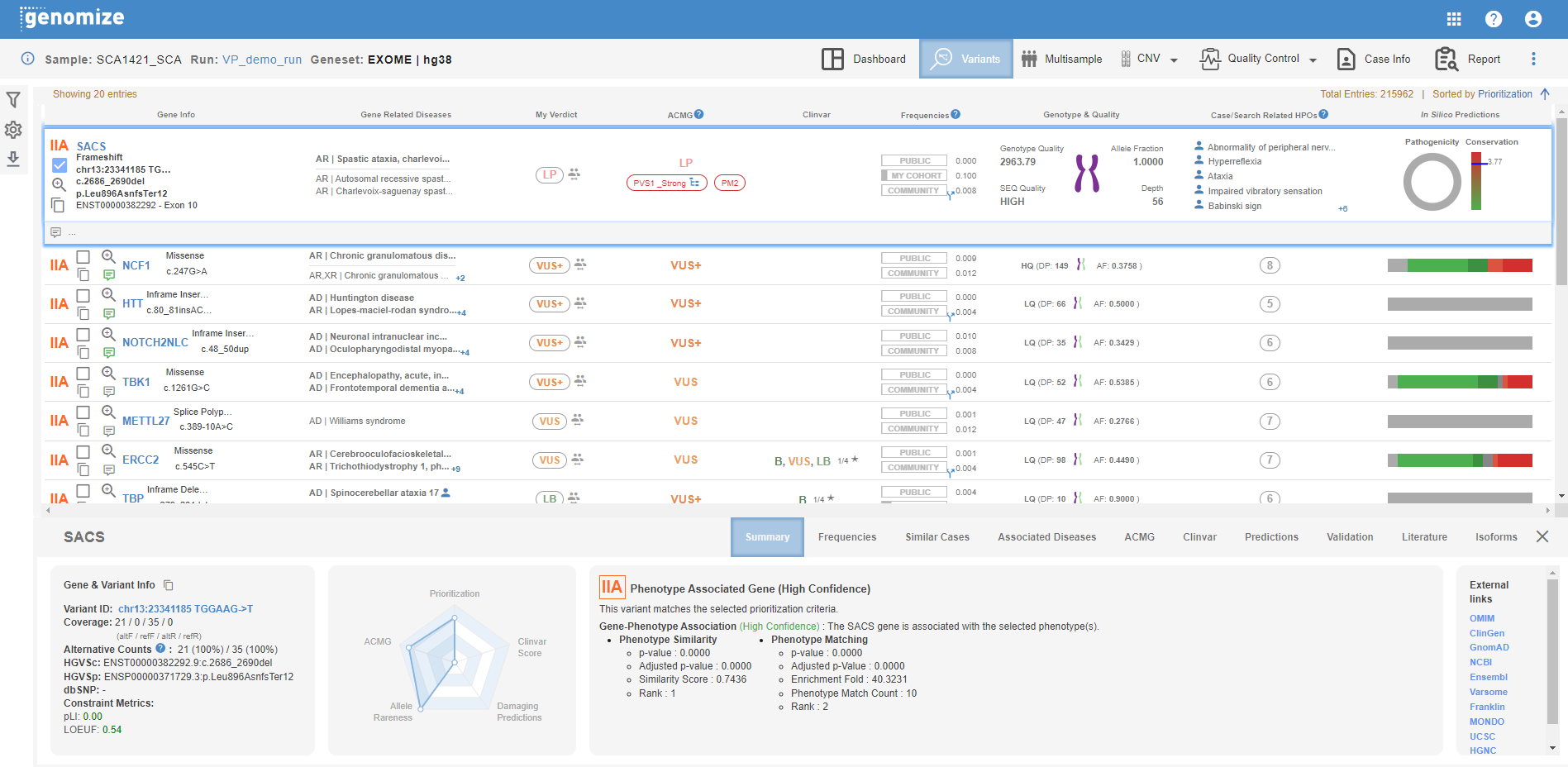
Here, you will see the information required for the variant evaluation in various columns as well as more detailed information about the gene and the variant in the summary area at the bottom of the page.
Gene Info: This column will list the gene name, the effect of the variant on the protein, changes in the transcript and the protein, as well as the chromosome of the gene and the affected exon.
- Variant Prioritization tier is shown here.
- You can use the checkbox to include the variant in your report.
- You can use the magnifying glass icon for IGV visualization.
- You can use the "Copy" button to copy the variant information to your computer's memory, and paste it using CMD + V or CTRL + V. You can change the content and order of the copied information on the user settings page.
Related Diseases: The name and MOI of the disease(s) associated with the gene are listed here. If there are more than 5 entries, you will see a "+x" icon at the bottom right to give you the total number of listed diseases. You can mouse over the list to see the complete list.
My Verdict: The SEQ Platform allows you to override the ACMG prediction. You can change your own ACMG classification by clicking on the "ACMG classification" in this column. If you change the classification, this change will be saved in your center's database, and you and your colleagues will see your choice in your future samples. User name and the date for the latest change is shown below the classification. You will also see a "people" icon on the right of this field. If any other center in the SEQ community makes a change in the ACMG classification of this variant, you will be able to see a concise list of the number of other centers that have changed the pathogenicity of this variant, and to which classification.
ACMG: Here, you will see Genomize's ACMG classification for the variant as well as the assigned evidence codes.
ClinVar: This column shows the ClinVar entries and the star status of the variant.
Frequencies: The frequency of the variant in "public" databases (i.e. GnomAD, ESP6500, UK10K, etc.), in your patient cohort, and the SEQ Community cohort, respectively. You will also see a "diverged arrow" icon if there is an equivalent variant in the hg19 genome version. When you mouse over this icon, you will see the equivalent variant's frequency information as well.
Genotype and Quality: Here, you will see the visual representation of the variant's genotype as well as various quality information. The compound heterozygous variants are represented with a "shadowed" chromosome image. Clicking on the chromosome image will list the compound heterozygous variants in the sample. For ONT and PacBio samples, compound heterozygosity uses phasing information and will show only trans variants.
- Genotype Quality: The quality value assigned by the variant caller used in your analysis.
- SEQ Quality: If the number of reads of the variant is between the primary and secondary coverage thresholds (5 and 50 by default, respectively), the variant will be classified as LOW quality. If it is higher than the secondary coverage threshold, it will be classified as HIGH quality. The threshold values can be changed during the sample upload or through the "Site settings".
- Allele Fraction: The ratio of the reads containing the variant to all reads covering the variant's position.
- Depth: The total number of reads covering the variant's position.
HPOs: You will see the phenotypes associated with the relevant gene in this column.
Computer Predictions: Here, you will see a horizontal bar graph in a collapsed view, a donut chart, and a vertical bar graph in a detailed view. The bar graph in the collapsed view and the donut chart in the detailed view show the predictions from 25 in-silico damage prediction tools, if available. The vertical bar graph shows the evolutionary conservation score prediction for the variant's position, if available.
Variant Notes: You can take notes in this field specific to the variant. Your notes will be saved in your center's database, and you and your colleagues will see these notes in your future samples. User name and the date for the last changes is shown below the the notes.
# Information Field
When you click on a variant card, the information field will pop up at the bottom of the page. In this field, you will see several tabs:
- Summary: This tab contains summary gene & variant information, a radar plot visualizing the pathogenicity and the relevance of the variant according to 5 categories, detailed information about the variant's classification in Genomize's AI-assisted variant prioritization feature, and external links for the variant and/or the gene.
- Frequencies: Here, you will see the frequency tables for the variant within your cohort, the SEQ community cohort, and the public databases:
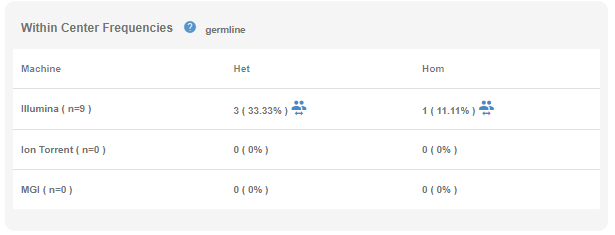
- Within Center Frequencies: In this table, you will see the frequency values as well as the incidences of the variant among your own sample cohort stratified by the sequencing platform and genotype. If the variant is encountered in other samples, you will see a "people" icon next to the frequency information. By clicking this icon, you can view the list of samples that carry this variant, and click on the sample name to open the sample in a new tab.
- SEQ Frequency: In this table, you will see the same information but for the whole SEQ community. Joining the SEQ community is optional and you can opt out anytime you like. If the variant has been encountered by any other center in the SEQ community, you will see a "hospital" icon next to the frequency information. You can see the number of other centers, and by clicking on this icon, you can get in contact with the other centers. You will not be able to obtain any information about the name of the center and the recipient unless the receiving party accepts your request.
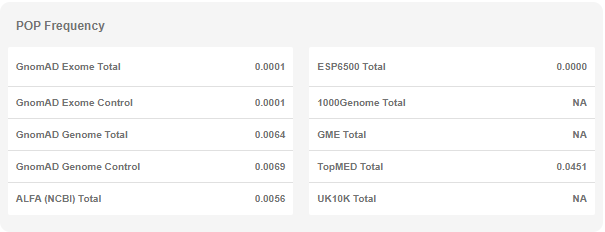
- Population Frequencies: This table shows the summary frequency values in various public databases. You can see the stratified values for each database in the "Variant Page" (see below).
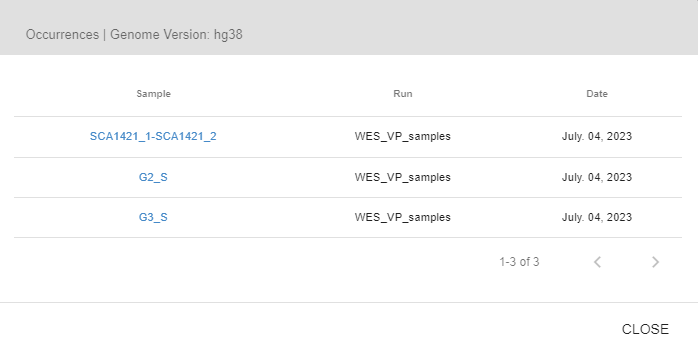
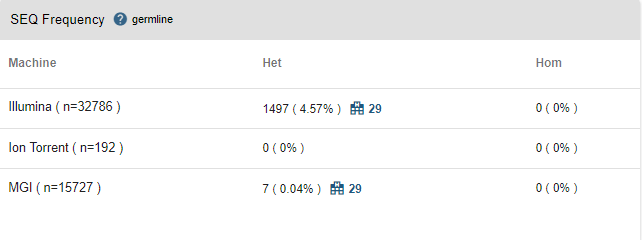
Similar Cases: In this tab, you will see the phenotypes observed in samples that carry this variant. The phenotype does not need to be observed in the sample you are currently analyzing. You can also see the number of samples with the specified phenotype as well as the names and links to those samples.
ACMG: Here, you can obtain more information about the ACMG classification of the variant. You can also use this tab as an ACMG calculator by adding, removing, or modifying the evidence codes.
ClinVar: In this tab, you will see the ClinVar entries for the variant and relevant links.
Predictions: Here you will see prediction scores from various in-silico tools for pathogenicity, evolutionary conservation, and splicing.
Validation: This form can be used to keep track of verifications (ex: Sanger or qPCR) for the variant.
Literature: You will see the list of publications for the variant and the gene listed separately. LitVar2 is used for variant literature. PubMed, MedGene and NCBI-Gene are used for publications containing the gene of interest. You can toggle the "disease-related only" switch to list all publications or only disease-related publications. You can also find the number of publications containing the gene in Mastermind and the link to the relevant page. This literature search is performed at the time of sample upload.
- Isoforms: All transcript annotations for the variant are listed in this table. The reference transcripts will appear on top with the default sorting. Please see the "Extended Annotation" section for more details.
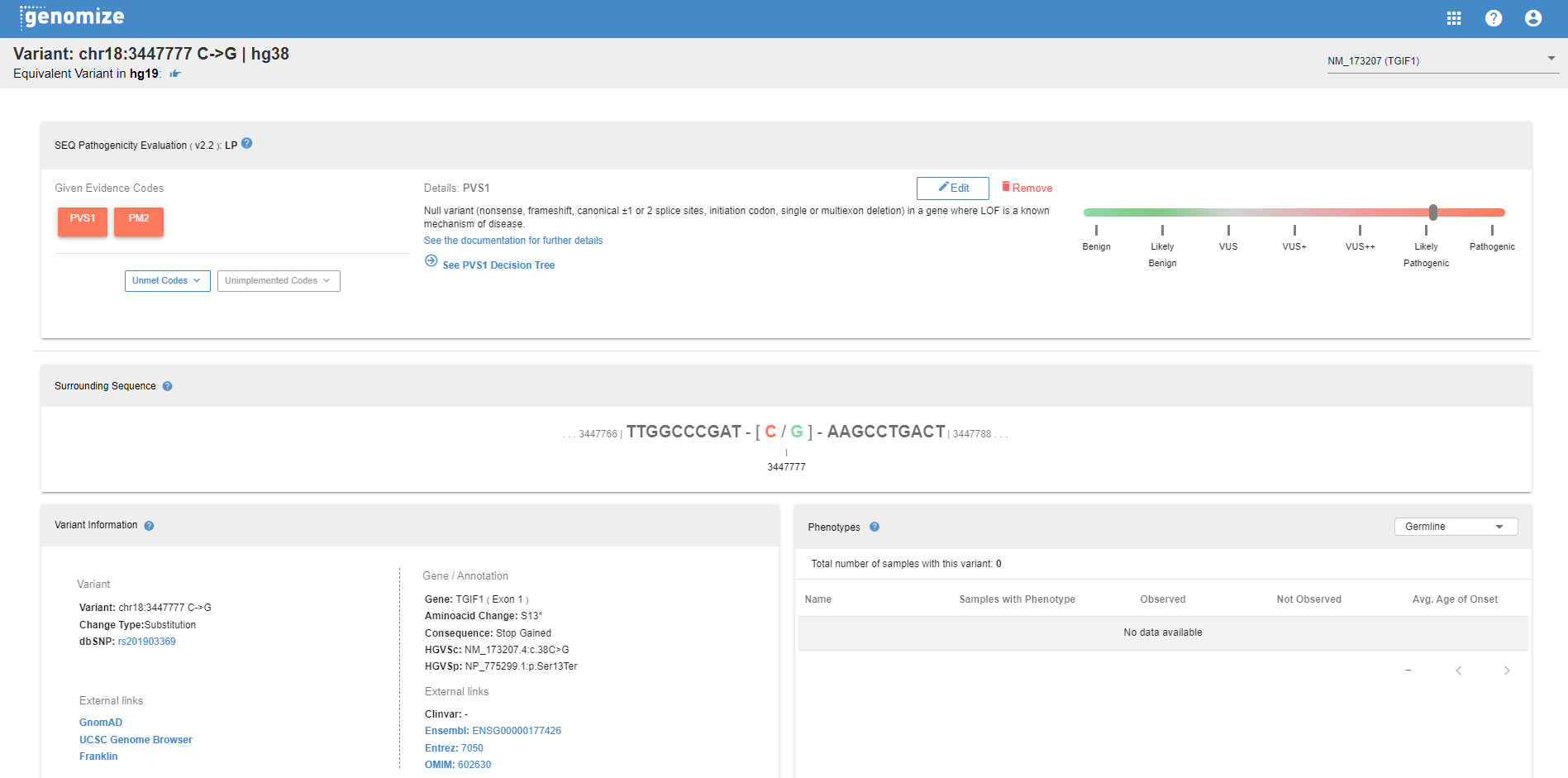
# The Variant Page
When you click on the "Variant ID" in the "Summary" tab of the information field, the variant page will open in a new tab. On this page, you can access various information most of which is available in the information field.
- ACMG pathogenicity calculator: Users can alter the evidence codes assigned for the variant by the SEQ.
- Surrounding Sequence: The genomic sequence around the variant is shown here.
- Variant Information: Basic variant information along with links to other important sources are listed.
- Phenotypes: The phenotype information you enter through the phenotypes section of every sample will be allocated in this section.
- Clinvar Hotspots: Pathogenic and Likely pathogenic variant distribution in the ClinVar database is shown together with the gene structure and domains from the UniProt database
- Transcripts: All transcript annotations for the variant are listed in this table. The reference transcripts will appear on top with the default sorting.
- Clinvar Entries: Clinvar entries are summarized.
- Population Frequencies: Population frequencies for various databases are listed in this table. You can see the details for subgroups and various stratifications here.
- Center/Community Frequencies: Genotype frequencies segregated by technology are illustrated.
# Filtering
The SEQ Platform offers various filtering options. You can open the filter options by clicking the "funnel" icon located at the top-left of the page.
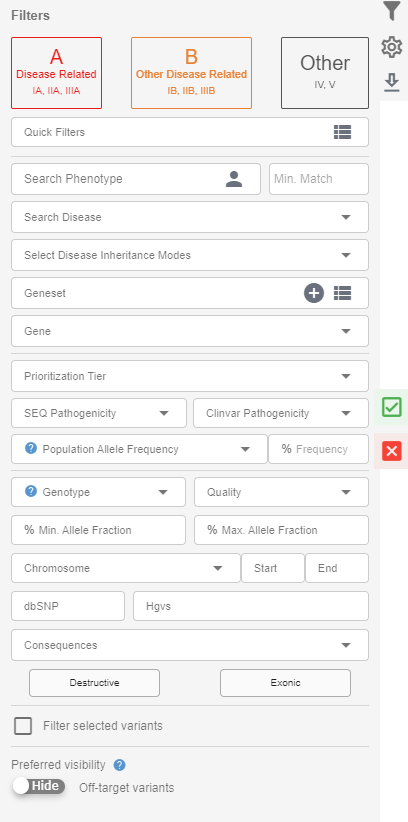
In the "Filters" field, you will see the following:
- Quick filter buttons for class "A", "B" and "others" variants according to the AI-assisted variant prioritization classification. Please refer to the relevant section of the help page and our white paper (opens new window) for more information.
- Quick filters: You can select a filterset you previously created for a quick application. You can create a new filterset by clicking the icon on the right of the "Quick Filters" section.
- Phenotypes: You can add phenotypes to your search by typing HPO phenotypes in this field. You can also use the shortcut in this field (person icon) to quickly add the phenotypes you added during the upload. You need to provide a number in the "Min. Match" field. This number will be used as the threshold for the matching phenotypes. For example, if you add five phenotypes to the search field and choose only three as the minimum match threshold, any matches with at least the three phenotypes in the list will be shown.
- Disease: You can perform a search for disease-gene associations.
- Geneset: You can create your own geneset using the plus icon in this field and use it in your filters. You can create as many genesets as you need. You can also use the system-defined genesets, which are updated weekly, or as the new version is released in the case of ACMG's list of recommended genes for incidental findings.
- Gene: You can search for variants in the gene(s) you want in using this option. You can search for multiple genes.
- Prioritization tier: This section will allow you to filter for variants with specific variant prioritization categories.
- SEQ pathogenicity: You can select the ACMG classification by the SEQ Plaform to filter for variants in this section.
- ClinVar pathogenicity: You can use ClinVar's variant classification categories for your filtering in this section.
- Population allele frequency: You can search for variants with higher/lower fractions in the public databases than the value you provide here. The population frequency filter will consider the GnomAD exome frequencies if the variant is covered in the GnomAD. The maximum allele frequency from the Exac, Esp6500, and 1000genome total frequencies will be used only if the variant is not covered in GnomAD. The reason for this order is due to the presence of possible ethnicity biases in the Esp6500, Exac, and 1000genome which are less likely to be present in the GnomAD since GnomAD contains many more samples from an evenly distributed sample set in terms of ethnicity. For instance, some well-established cystic fibrosis variants are present in more than 5% of the ESP6500 EA population.
- Genotype: Filter for het or hom variants.
- Quality: You can filter for high- or low-quality variants. Please see above for the "SEQ Quality".
- % Min. allele fraction & % max. allele fraction: You can provide % values in these fields to filter for variants within the range of these values.
- Chromosome, start and end: You can search for variants within the genomic coordinates you provide in this section.
- dbSNP: You can search for a specific variant using the rdID here.
- HGVS: You can search for a specific variant using the HGVS identifier here.
- Consequences: You can select the effect of the variant on the protein/transcript here. You will see two shortcut buttons for "Destructive" and "Exonic" effects.
- Filter selected variants: If an option is selected, only the variants that have the "report checkbox" ticked will be used for filtering.
- Preferred visibility: You can toggle the visibility of off-target and off-geneset (not available for WES samples) here. You can make this change permanent on the account settings page.
Please note that the filters work with the "AND" operator.
# ACMG Classification
You can see the explanation of Genomize for the principles and thresholds used for a particular evidence code by clicking on that evidence code. Find below the details of each evidence code based on the autopathogenicity version of the analysis.
# Evidence Codes (Autopathogenicity v2.x)
# PVS1
A Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where the LOF is a known mechanism of a disease.
Null variant types:
- Stop gained/Frameshift (Nonsense or Frameshift variant)
- Splice acceptor/donor (GT-AG, 1,2 splice sites)
- Start lost (Initiation Codon)
- Deletion (Inframe or full gene deletion)
Loss of function is a mechanism of the disease if (either of the below is required):
- At least 3 pathogenic LoF variants from the ClinVar (with a minimum of 1 star) need to be present
- The LOEUF score of the transcript is smaller than 0.35 as suggested by GNOMAD
The transcript is biologically relevant if it is present in:
- All RefSeq transcripts
- Ensembl transcripts with expression in any of the 51 GTeX (v6) tissues (excluding expression data from two cell types)
The LoF variant in a particular exon is frequent in the general population if:
- The 99th percentile of the allele frequency distribution of the previously reported LoF variants (GnomAD) in the exon is higher than the predefined threshold of 0.1%.
The Truncated/Altered region is critical for the protein function if:
- For the nonsense/frameshift mutations: The altered region is the region between the variant position and the last base of the coding sequence (excluding the stop codon).
- For the splice mutations: If the ORF is preserved, we consider the nearby (upstream for splice donor, downstream for splice acceptor) skipped exon as the altered region. Otherwise, it is the region between the variant position and the last base of the coding sequence.
- For the deletion mutations: If the ORF is preserved, we consider only the skipped region of the protein. Otherwise, it is the region between the variant position and the last base of the coding sequence.
- For the altered region to be critical for the protein, we require at least 3 pathogenic ClinVar variants with a minimum of 2 stars (multiple submitters) or higher.
Expected to undergo NMD (nonsense-mediated decay) if:
- The variant is not present in the 3' most coding exon or the 3' most 50 bp of the penultimate coding exon.
- The transcript has a multi-coding exon.
# PS1
The same amino acid change as a previously established pathogenic variant regardless of the nucleotide change.
Evidence code strength is modified based on the Clinvar review status:
- 1-star: PS1 (Supporting)
- 2-star: PS1 (Moderate)
- 3-4 star: PS1 (Strong)
# PM1
Located in a mutational hotspot and/or critical and well-established functional domain (e.g., an active site of an enzyme) without benign variation.
Detection of a mutational hotspot
A hotspot region is defined as the largest pathogenic variant dense region between two benign variations and meeting the following conditions:
- Presence of a minimum of 5 pathogenic and 0 benign variants reported in the ClinVar.
- Pathogenic variant count per base pair of the region is greater than the predefined threshold of 0.2.
Functional UniProt domain
To define a functional UniProt domain, the following rules are employed:
- UniProt domains with ( 'chain', 'coiled-coil region', 'transit peptide', 'helix', 'turn', 'beta strand') categories are excluded.
- Domains without benign variation in the ClinVar (with a minimum of 1 star).
- At least 3 pathogenic variants in the ClinVar (with a minimum of 1 star).
# PM2
Absent from controls (or at extremely low frequency if recessive) in the Exome Sequencing Project, the 1000 Genomes Project, or the Exome Aggregation Consortium databases.
- Maximum allele frequency of the variant is less than the predefined threshold.
# PM4
The protein length changes as a result of in-frame deletions/insertions in a non-repeat region or stop-loss variants.
# PM5
A novel missense change at an amino acid residue where a different missense change was previously observed as a pathogenic variant.
Evidence code strength modified based on the Clinvar review status:
- 1-2 star: PM5 (Supporting)
- 3-4 star: PM5 (Moderate)
# PP2
A missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease.
This rule only applies to missense variants. The following thresholds are applied to determine if a missense variant is a common mechanism of disease:
- Minimum missense pathogenic variant count in the transcript: 5
- Missense/total pathogenic variant count ratio threshold: 70%
- Missense pathogenic/benign count threshold: 0.65
# PP3
Multiple lines of computational evidence support a deleterious effect on the gene or the gene product (conservation, evolutionary, splicing impact, etc.).
PP3 is assigned if one the following criteria is met (Ionnadis et al. 2016 (opens new window)):
- The REVEL score is greater than 0.75
- The REVEL score is greater than 0.5 and MetalR is damaging
- The REVEL score is not available and MetalR is damaging
# PP5
A reputable source recently reported the variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation.
Evidence code strength is modified based on the Clinvar review status:
- 1-star: PP5 (Supporting)
- 2-star: PP5 (Moderate)
- 3-star: PP5 (Strong)
- 4-star: PP5 (Very strong)
# BA1
The allele frequency is >5% in the Exome Sequencing Project, the 1000 Genomes Project, or the Exome Aggregation Consortium databases.
- Maximum allele frequency of the variant is greater than 5%
- Variants on the BA1 exception list provided by Clingen are excluded. Clingen Exception List (opens new window)
# BS1
The allele frequency is greater than the expected frequency for the disorder.
- Maximum allele frequency of the variant is less than the predefined BS1 threshold
# BS2
Observed in a healthy adult individual for a recessive (homozygous), dominant (heterozygous), or X-linked (hemizygous) disorder, with full penetrance expected at an early age.
- A variant was observed (at least 2 times) in the control cohort of the Gnomad (v2.1.1) database in a homozygous state.
# BP1
A Missense variant in a gene for which primarily truncating variants are known to cause disease.
- At least 5 pathogenic variants are reported in the ClinVar database.
- All pathogenic variants in the ClinVar database are truncating.
# BP3
In-frame deletions/insertions in a repetitive region without a known function.
BP3 is assigned if the variant is in a repetitive region and the repetitive region meets the following criteria.
- Do not overlap with a functional UniProt domain.
- Do not have a pathogenic variant reported in the ClinVar.
# BP4
Multiple lines of computational evidence suggest no impact on the gene or the gene product (conservation, evolutionary, splicing impact, etc.).
BP4 is assigned if one the following criteria is met (Ionnadis et al. 2016 (opens new window)):
- The REVEL score is less than 0.5
- The REVEL score is less than 0.75 and MetalR is benign
- The REVEL score is not available and MetalR is benign
# BP6
A Reputable source recently reported the variant as benign, but the evidence is not available to the laboratory to perform an independent evaluation.
Evidence code strength is modified based on ClinVar review status:
- 1-2-star: BP6 (Supporting)
- 3-star: BP6 (Strong)
- 4-star: BP6 (Stand alone)
# BP7
A synonymous (silent) variant for which splicing prediction algorithms predict no impact to the splice consensus sequence nor the creation of a new splice site AND the nucleotide is not highly conserved.
BP7 is assigned if the following criteria are met:
- Cryptic splice site is not predicted (ADA and RF scores are less than 0.6)
- Low conservation score (phylop100way score is less than 1.879)
# Unimplemented Evidence Codes
The following evidence codes are unimplemented: PS2, PS3, PS4, PM3, PM6, PP1, PP4, BS3, BS4, BP2, BP5.
# Evidence Codes (Autopathogenicity v1.x)
# PVS1
A Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where LOF is a known mechanism of a disease.
Null variant types:
- Stop gained / Frameshift (Nonsense or Frameshift variant)
- Splice acceptor/donor (GT-AG, 1,2 splice sites)
- Start lost (Initiation Codon)
The LoF (Loss of function) is classified as a mechanism of disease if:
- At least 1 pathogenic LoF variant from the ClinVar (with a minimum of 1 star) needs to be present.
Expected to undergo NMD (nonsense-mediated decay) if:
- The variant is not present in the 3' most coding exon or the 3' most 50 bp of the penultimate coding exon.
- The Transcript has multi-coding exon.
# PS1
The same amino acid change as a previously established pathogenic variant regardless of nucleotide change.
Evidence code strength modified based on the Clinvar review status.
- 1-star: PS1 (Supporting)
- 2-star: PS1 (Moderate)
- 3-4 star: PS1 (Strong)
# PM1
Located in a mutational hotspot and/or critical and well-established functional domain (e.g., an active site of an enzyme) without a benign variation.
Detection of a mutational hotspot
A hotspot region is defined as the largest pathogenic variant dense region between 2 benign variations and meeting the conditions below:
- Presence of a minimum 5 pathogenic and 0 benign variants reported in the ClinVar.
- The Pathogenic variant count per base pair of the region is greater than the predefined threshold of 0.2.
Functional UniProt domain
To define a functional UniProt domain the following rules are employed:
- UniProt domains with ( 'chain', 'coiled-coil region', 'transit peptide', 'helix', 'turn', 'beta strand' ) categories are excluded.
- Domains without benign variation in the ClinVar (with a minimum of 1 star)
- At least 3 pathogenic variants in the ClinVar (with a minimum of 1 star)
# PM2
Absent from controls (or at extremely low frequency if recessive) in the Exome Sequencing Project, the 1000 Genomes Project, or the Exome Aggregation Consortium databases.
- Maximum allele frequency of the variant is less than the predefined threshold of 5%
# PM4
The protein length changes as a result of in-frame deletions/insertions in a non-repeat region or stop-loss variants.
# PM5
A novel missense change at an amino acid residue where a different missense change was previously observed as a pathogenic variant.
Evidence code strength modified based on the Clinvar review status.
- 1-2 star: PM5 (Supporting)
- 3-4 star: PM5 (Moderate)
# PP2
A missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease.
This rule only applies to missense variants. The following thresholds are applied to determine if a missense variant is a common mechanism of disease:
- Minimum missense pathogenic variant count in the transcript: 5
- Missense/total pathogenic variant count ratio threshold: 70%
- Missense pathogenic/benign count threshold: 0.65
# PP3
Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.
PP3 is assigned if one the following criteria is met (Ionnadis et al. 2016 (opens new window)):
- REVEL score is greater than 0.75
- REVEL score is greater than 0.5 and MetalR is damaging
- REVEL score is not available and MetalR is damaging
# PP5
A reputable source recently reported the variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation.
Evidence code strength is modified based on the Clinvar review status:
- 1-star: PP5 (supporting)
- 2-star: PP5 (moderate)
- 3-star: PP5 (strong)
- 4-star: PP5 (very strong)
# BA1
The allele frequency is >5% in the Exome Sequencing Project, the 1000 Genomes Project, or the Exome Aggregation Consortium databases.
- Maximum allele frequency of the variant is greater than 5%
# BS2
Observed in a healthy adult individual for a recessive (homozygous), dominant (heterozygous), or X-linked (hemizygous) disorder, with full penetrance expected at an early age.
- A variant was observed (at least 2 times) in the control cohort of the Gnomad (v2.1.1) database in a homozygous state.
# BP1
A Missense variant in a gene for which primarily truncating variants are known to cause disease.
- At least 5 pathogenic variants are reported in the ClinVar database.
- All pathogenic variants in the ClinVar database are truncating.
# BP3
In-frame deletions/insertions in a repetitive region without a known function.
BP3 is assigned if the variant is in a repetitive region and the repetitive region meets the following criteria.
- Do not overlap with a functional UniProt domain.
- Do not have a pathogenic variant reported in the ClinVar.
# BP4
Multiple lines of computational evidence suggest no impact on the gene or the gene product (conservation, evolutionary, splicing impact, etc.).
BP4 is assigned if one the following criteria is met (Ionnadis et al. 2016 (opens new window)):
- REVEL score is less than 0.5
- REVEL score is less than 0.75 and MetalR is benign
- REVEL score is not available and MetalR is benign.
# BP6
A Reputable source recently reported the variant as benign, but the evidence is not available to the laboratory to perform an independent evaluation.
Evidence code strength is modified based on ClinVar review status:
- 1-2-star: BP6 (Supporting)
- 3-star: BP6 (Strong)
- 4-star: BP6 (Stand alone)
# Unimplemented Evidence Codes
The following evidence codes are unimplemented: PS2, PS3, PS4, PM3, PM6, PP1, PP4, BS1, BS3, BS4, BP2, BP5, BP7.